










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSAMJ: South African Medical Journal
On-line version ISSN 2078-5135
Print version ISSN 0256-9574
SAMJ, S. Afr. med. j. vol.106 n.3 Pretoria Mar. 2016
http://dx.doi.org/10.7196/samj.2016.v106i3.9476
RESEARCH
A clinical and molecular investigation of two South African families with Simpson-Golabi-Behmel syndrome
C SpencerI, II; K FieggenIII; A VorsterIV; P BeightonV
IMB ChB, DCH, FCMG, MMed (Genet); Division of Human Genetics, Faculty of Health Sciences, University of Cape Town, South Africa
IIMB ChB, DCH, FCMG, MMed (Genet); Division of Human Genetics, School of Pathology, Faculty of Health Sciences, University of the Witwatersrand, Johannesburg, South Africa, and National Health Laboratory Service, Johannesburg
IIIMB ChB, FCPaed (SA), Cert Med Genet; Division of Human Genetics, Faculty of Health Sciences, University of Cape Town, South Africa
IVMSc; Division of Human Genetics, Faculty of Health Sciences, University of Cape Town, South Africa
VOMB, MD, PhD, FRCP, FRSSA; Division of Human Genetics, Faculty of Health Sciences, University of Cape Town, South Africa
ABSTRACT
BACKGROUND: Simpson-Golabi-Behmel syndrome (SGBS) is an X-linked recessive overgrowth syndrome manifesting primarily in boys and characterised by macrosomia, distinctive facial features and multiple congenital abnormalities. Although this rare condition is thought to be underdiagnosed, making a diagnosis is important as affected boys have a 7.5% risk of developing visceral tumours and surveillance is warranted. Mutations in GPC3 are found in up to 70% of boys affected with SGBS.
OBJECTIVES: A clinical and molecular investigation of two boys with SGBS, probands B and S, and their mothers. Documentation of the clinical phenotype could assist with diagnosis in affected boys and will lead to early initiation of tumour surveillance.
METHODS: Hospital folders were reviewed and clinical consultations arranged for both probands and their mothers. Molecular investigations initially searched for whole-exon deletions in GPC3 followed by gene sequencing.
RESULTS: The clinical phenotype of both probands was consistent with that previously reported in the literature. The main features pointing towards the diagnosis were macrosomia, coarse facial features and macroglossia with a midline groove in the tongue. Proband B developed a Wilms tumour. He was found to have a novel mutation causing a premature stop codon.
CONCLUSIONS: This research represents the first published report of SGBS in South Africa. Early recognition and confirmation of this condition is important in order to institute tumour surveillance and assist families with accurate recurrence risks.
Simpson-Golabi-Behmel syndrome (SGBS) (OMIM #312870) is an X-linked overgrowth syndrome characterised by distinctive facial features, macro-somia and multiple congenital abnormalities.[1] This is a rare condition, with only about 100 molecularly confirmed cases reported in the literature.[2] The clinical picture can be difficult to distinguish from other overgrowth conditions, with which it shares many overlapping features.[3] This could lead to misdiagnosis and therefore under-reporting of SGBS. There have been no previous published reports of this condition in South Africa (SA).
SGBS was originally delineated by Simpson et al.[4]in 1975, who described boys with 'bulldog-like' facies. However, it was only in 1988 that Neri et al.[5] coined the eponymous term 'Simpson-Golabi-Behmel syndrome', after the original author and those of two subsequent articles describing the same phenotype.
Boys with SGBS have pre- or postnatal macrosomia. Their facial features are coarse, with hypertelorism, downslanted palpebral fissures and a broad nasal bridge.[1] They also have a prominent jaw, with macrostomia and macroglossia. A groove in the lower lip or tongue is frequently present. The nose is often short and upturned and about 25% have a cleft lip or palate.[6] Polydactyly with short, broad hands has been described.[7]
Other congenital abnormalities include cardiac defects in 36% of affected boys[8] and skeletal problems such as pectus excavatum, scoliosis and vertebral abnormalities. Gastrointestinal and renal abnormalities can also occur.[1] Common urogenital malformations are undescended testes and inguinal hernias, but hypospadias and ambiguous genitalia have been reported.[9] The intellectual outcome is variable, with some individuals having normal development and others exhibiting developmental delay. Speech delay is diagnosed in about 50% of affected boys and motor delay in 36%.[6] Hypotonia is consistently present and structural brain abnormalities can occur.[6]
Importantly, boys with SGBS have an approximately 7.5% risk of developing tumours. The spectrum of tumours observed includes Wilms tumour, hepatoblastoma, neuroblastoma, gonadoblastoma, hepatocellular carcinoma and medulloblastoma.[10] It is widely accepted that this risk is sufficient to warrant regular tumour surveillance.
SGBS is inherited in an X-linked recessive manner. Males manifest the condition, whereas females are usually asymptomatic carriers. However, females can exhibit some features such as tall stature, coarse facies and occasionally intellectual disability.[1]
Two adjacent genes on the X chromosome, GPC3 and GPC4, have been implicated in SGBS. GPC3 was the first gene described in patients with SGBS. Mutations in this gene have been found in up to 70% of people with the syndrome.[7,11] There are no known hotspots, and a variety of mutations ranging from deletions to point mutations can occur in any of the eight exons.[11] Recently, exon duplications have also been reported.[2] Intragenic GPC4 mutations have not been described in isolation and are usually an extension of a deletion that includes GPC3. While there has been a report of a GPC4 duplication causing SGBS in a family,[12] this duplication cannot explain the mechanism of disease. In subsequent studies no other mutations in GPC4 have been identified.[2]
Objectives
A clinical and molecular evaluation of two probands with SGBS. A description of the phenotype could assist medical professionals in SA to make the diagnosis, which in turn would lead to early initiation of tumour surveillance. Molecular testing can not only confirm a clinical diagnosis but also offer the option of carrier and prenatal testing to family members at risk.
Methods
Ethics approval was received from the Human Research Ethics Committee of the Faculty of Health Sciences, University of Cape Town, SA (HREC REF 072/2012). Two boys with a clinical diagnosis of SGBS attending the genetics clinic at Red Cross War Memorial Children's Hospital in Cape Town were identified. These probands were designated B and S. Their hospital folders were reviewed for history, clinical findings and special investigations. Clinical consultations were arranged for both boys along with their mothers, and pertinent clinical features were documented.
The molecular analysis was undertaken in the Division of Human Genetics, University of Cape Town. DNA was extracted from venous blood specimens for both probands and their mothers using standard methods. This DNA was analysed for whole-exon deletions and thereafter for sequence abnormalities in GPC3.
The primers selected for the amplification of GPC3 coding regions were those reported in 2007 by Sakazume et al.[13] Exon 3 was analysed with two overlapping sets of primers owing to the large size of the coding region.
The exons were amplified in singleplex using the polymerase chain reaction (PCR) in a 25 μL. final reaction volume consisting of 100 ng template DNA, 10 pmol of each primer, 200 μΜ of each of the deoxynucleotide triphosphates (Bioline, UK) and 0.5 units of GoTaq DNA polymerase (Promega, USA). Following amplification of this reaction on the BioRad T100 thermal cycler (BioRad Laboratories, USA), the yield and specificity of the resulting fragments were confirmed by electrophoresis on a 1.5% (w/v) agarose gel prior to direct cycle sequencing.
Sequencing for both probands and their respective mothers was performed using primers employed for the PCR, with subsequent capillary electrophoresis on the 3130xl Genetic Analyser (Life Technologies, USA). The results of the sequencing reaction were analysed using SeqMan (DNASTAR, USA) software systems. The mutation found was investigated using two databases, the Leiden Open Variation Database[14] and the National Center for Biotechnology Information website.[15]
Results
Clinical features Proband B
Proband B was evaluated at the age of 5 years and 9 months. There was no family history of intellectual concerns, although a maternal cousin had a cardiac lesion and bowel problem. No further details were known. The boy is his mother's only child.
Proband B was born prematurely by vaginal delivery in the breech position. His birth weight was 2 100 g (>90th centile) and his head circumference 31 cm (>90th centile). Postnatally he required ventilation for aspiration pneumonia and had prolonged jaundice and a single episode of hypoglycaemia.
Proband B demonstrated persistent overgrowth, all his growth parameters consistently measuring above the 90th centile with some acceleration of his height and head circumference noted. On examination proband B had a long face with hypertelorism, epicanthic folds and an upturned nose (Fig. 1). His nasal bridge was broad. His mouth tended to be open and he had macrostomia and macroglossia with a grooved tongue and prognathism. He also had pectus excavatum and a marked lordosis.

He had an atrioseptal defect with pulmonary valve stenosis. Genitourinary malformations including bilateral inguinal hernias, undescended testes and hypospadias were noted at birth. He was subsequently found to have right-sided hydronephrosis secondary to pelvoureteric obstruction. At ~6 years of age he was diagnosed with a Wilms tumour of the left kidney. His developmental milestones had all been within the normal range, but he had been noted to be hypotonic. At the initial review his speech was still slightly difficult to comprehend, but the rest of his development was appropriate for his age.
Additional investigations revealed a normal male karyotype (46,XY) and 13 pairs of ribs on the chest radiograph.
Proband B's mother was a healthy woman of normal intellect. She had been diagnosed with scoliosis during adolescence, which resolved spontaneously. She was tall, and at 1.81 cm her height plotted above the 97th centile. She had a long face with no other obvious dysmorphic features.
Proband S
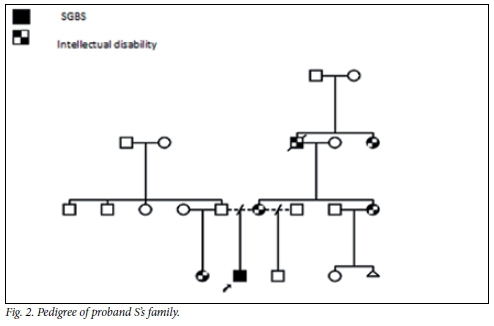
Proband S was evaluated at 4 years and 2 months of age. As shown in the pedigree (Fig. 2), mild intellectual disability was present in the family. His mother has asthma, but was otherwise in good health during the pregnancy.
Proband S had been born via normal vertex delivery at term. He had Apgar scores of 8 and 9. His birth weight was 3 200 g (10th centile) and his head circumference 33 cm (5th centile). Postnatally he was ventilated for apnoea and had phototherapy for jaundice.
At the time of examination, proband S's weight was above the 50th centile, his height above the 75th centile and his head circumference above the 95th centile, reflecting growth acceleration. He had a coarse face with hypertelorism, downslanted palpebral fissures, epicanthic folds and an upturned nose (Fig. 3). His tongue was protruding, macroglossic and had a central groove. He also had macrostomia. He was mildly hirsute with a low posterior hairline.

Proband S had broad, short fingers with bilateral fifth-finger clinodactyly. His toes were broad and he had pes planus. He had mild lumbar lordosis. He had a patent ductus arteriosus with a patent foramen ovale that closed spontaneously. There were no genitourinary abnormalities. Abdominal examination showed hepatomegaly of 2 cm below the costal margin, which had been stable for several years. He was hypotonic and had a developmental quotient of 60, in keeping with mild intellectual disability.
Magnetic resonance imaging of his brain at 2 years of age showed atrophy and a peritrigonal white-matter high signal suggestive of a hypoxic insult at birth. A chest radiograph revealed 11 ribs on the right and 12 on the left. He had a normal male karyotype (46,XY).
At the time of examination, proband S had not developed any tumours and was attending a special educational needs school.
Proband S's mother had mild intellectual disability. She was functionally independent and in employment. Her height was 176 cm (above the 97th centile). She had a long face, with a high-arched palate. The other family members with intellectual disability were not available for examination.
Table 1 summarises the clinical features observed in the two probands and compares them with those frequently described in the literature.

Molecular investigations
No exonic deletions were found in GPC3 of proband B or proband S.
Sequencing revealed a mutation in exon 4 of GPC3 in proband B. This mutation consists of a deletion of four nucleotides, TAGA, at nucleotide position 1071, and an insertion of three nucleotides, CTT. This mutation, designated as c.1071_1074delinsCTT (Genbank accession No. NM_004484.3), causes a frameshift that results in a premature stop codon. At a protein level, this tetranucleotide deletion results in a disruption of the amino acid reading frame that causes arginine to be replaced by phenylalanine at position 358, with a resulting premature stop codon at position 373, p.(Arg358Phefs*16). As this premature stop codon occurs in exon 4, the majority of the coding sequence of GPC3 is not translated. This mutation was confirmed to be present in proband B's mother in the heterozygous state.
No sequence abnormalities were found in proband S or his mother.
Discussion
There are no published data describing SGBS in SA. The two boys in this study exhibited many of the more common features described in SGBS. A limitation of this study is that both of the probands investigated were of Caucasian ancestry.
The diagnosis of SGBS is not easy to make, and there is considerable overlap with the other overgrowth syndromes. Equally, the facial features may not be apparent in infants. In SA, where limited genetic testing for these syndromes is available, the clinician relies heavily on clinical presentation. Other genetic overgrowth conditions that need to be considered in the differential diagnosis of SGBS include Sotos syndrome and Weaver and Perlman syndromes. Although these syndromes share some clinical features, it is usually possible to distinguish between them. The condition that is most often confused with SGBS is Beckwith-Wiedemann syndrome (BWS). BWS is one of the more common overgrowth syndromes and is characterised by macrosomia, macroglossia and anterior abdominal wall defects. It is important to distinguish between these conditions, as they have different genetic aetiologies and prognoses. An X-linked inheritance pattern, rib malformations, nail hypoplasia, syndactyly of the second and third fingers and supernumerary nipples are indicative of a diagnosis of SGBS rather than BWS.[3] Omphaloceles are infrequently encountered in SGBS and are more suggestive of BWS. An additional distinguishing feature is that the clinical phenotype and facial features associated with SGBS tend to become more pronounced with age, whereas they tend to normalise in those affected with BWS.[3] Despite this, one should remain aware of the considerable overlap between these two conditions.
Both probands in this study were diagnosed with SGBS in early childhood. Macrosomia, coarse facial features, macro-glossia and a grooved tongue were the main clinical features leading to the diagnosis, supported by the cardiac and skeletal malformations in both boys and the genitourinary anomalies in proband B.
The variability in intellectual outcome is also evident in these boys. Proband S has a family history of mild intellectual disability. His mother and his maternal aunt are similarly affected and his maternal grandfather and great-aunt were reported to have intellectual difficulties too. This inheritance pattern could be consistent with X-linked inheritance, with females showing variable clinical expression. It is, however, not the only form of inheritance possible, and multifactorial causes for intellectual disability also warrant consideration.
Proband B developed a Wilms tumour. This tumour is known to be associated with SGBS. In a recent review of 63 boys with SGBS and a confirmed GPC3 mutation, only three had developed a Wilms tumour;[2] in this context, proband B represents the fourth published case.
Table 2 lists examinations and investigations recommended to monitor for tumour development.

Many different mutations in GPC3 have been described as causative in SGBS. No hotspots for these mutations have been identified, but frameshift mutations and deletions have been reported on numerous occasions. All mutations associated with SGBS either remove a start codon or introduce a premature stop codon. These premature stop codons invariably lead to a truncated protein with insufficient cysteine residues in the conserved cysteine motif.[11]
Although the specific mutation found in proband B has not previously been reported, its effect appears to be similar to other mutations associated with SGBS. The sequence anomaly disrupts the reading frame and introduces a premature stop codon, resulting in the majority of the protein not being translated. The databases investigated for this mutation confirmed that it is novel.
Proband B's mother was found to be heterozygous for this mutation. Accurate counselling regarding the recurrence risk for future pregnancies is now possible. In any future pregnancy, a male fetus will have a 50% chance of being affected with SGBS and a female fetus will have a 50% chance of being a carrier of this mutation. Antenatal diagnosis will be available to the mother should she want to pursue this option, following confirmation of the results in a diagnostic laboratory.
Testing of male offspring in infancy is another option. Testing of a minor would be ethically justified, as the result would have a direct impact on management with tumour surveillance. Cascade screening and genetic counselling have also been offered to proband B's family.
No mutation was identified in proband S, even though mutations in GPC3 are present in up to 70% of individuals with SGBS.'7,111 This could be because a gene other than GPC3 is responsible for his condition. GPC4 is such a candidate owing to the discovery of a duplication; however, since subsequent studies have failed to identify point mutations, other as yet unidentified genes should be considered. Equally, disruption of GPC3 could have been caused by a mechanism not examined in this study, such as splicing variations or intronic alterations. There have also recently been reports in the literature of exon duplications in GPC3 causing SGBS.[2] This abnormality would not have been identified by the methods used in this study and represents a limitation of this research. It is also possible that proband S does not have SGBS, but in fact another genetic condition. Nevertheless, based on his history and clinical features we believe that SGBS remains the most likely diagnosis.
Conclusions
This article represents the first published description of SA patients with SGBS, an underdiagnosed syndrome. The implications of a diagnosis of SGBS relate not only to the clinical diagnosis and recurrence risk in the family, but also to the predisposition for developing tumours.
Acknowledgements. This research was funded by the National Research Foundation of South Africa (PB). The authors thank Prof. R Ramesar, the genetic nurses and all the staff in the Division of Human Genetics, University of Cape Town, and in particular the patients and parents involved in the research.
References
1. Golabi M. Simpson-Golabi-Behmel syndrome type 1 -GeneReviews - NCBI Bookshelf 2011 (updated 23 June 2011). http://www.ncbi.nlm.nih.gov/books/NBK1219/ (accessed 24 August 2015). [ Links ]
2. Cottereau E, Mortemousque I, Moizard M, et al. Phenotypic spectrum of Simpson-Golabi-Behmel syndrome in a series of 42 cases with a mutation in GPC3 and review of the literature. Am J Med Genet C Semin Med Genet 2013;163C(2):92-105. [http://dx.doi.org/10.1002/ajmg.c.31360] [ Links ]
3. Knopp C, Rudnik-Schoneborn K, Zerres K, Gewncik M, Spengler S, Eggerman T. Twenty-one years to the right diagnosis - clinical overlap of Simpson-Golabi-Behmel and Beckwith-Wiedemann syndrome. Am J Med Genet 2015;167A(1):151-155. [http://dx.doi.org/10.1002/ajmg.a.36825] [ Links ]
4. Simpson JL, Landey S, New M, German J. A previously unrecognised X-linked syndrome of dysmorphia. Birth Defects Orig Art Ser 1975;11(2):18-24 [ Links ]
5. Neri G, Marini R, Cappa M, Borrelli P, Opitz JM. Simpson-Golabi-Behmel syndrome: An X-linked encephalo-tropho-schisis syndrome. Am J Med Genet 1988;30(1-2):287-299. [http://dx,doi.org/10.1002/ajmg/a.36317] [ Links ]
6. Mariani S, Iugheti L, Bertorelli R, Coviello D, Pelligrini M, Forabosco A. Genotype/phenotype correlations of males affected by Simpson-Golabi-Behmel syndrome with GPC3 gene mutations: Patient report and review of literature. J Pediatr Endocrinol Metab 2003;16(2):225-232. [http://dx.doi.org10.1515/JPEM.2003.16.2.225] [ Links ]
7. Neri G, Gurreiri F, Zanni G, Lin A. Clinical and molecular aspects of the Simpson-Golabi-Behmel syndrome. Am J Med Genet 1998;79(4):279-283. [http://dx.doi.org/10.1002/ajmg.a.36317] [ Links ]
8. Lin A, Neri G, Hughes-Benzie R, Weksberg R. Cardiac anomalies in the Simpson-Golabi-Behmel syndrome. Am J Med Genet 1999;83(5):378-381. [http://dx.doi.org10.1002/(SICI)1096-8628(19990423)83:53.3.CO;2-1] [ Links ]
9. Griffith C, Probert R, Vance G. Genital anomalies in three male siblings with Simpson-Golabi-Behmel syndrome. Am J Med Genet 2009;149A(11):2484-2488. [http://dx.doi.org/10.1002.ajmg.a.33047] [ Links ]
10. Yalchelevich N. Generalised overgrowth syndromes with prenatal onset. Curr Probl Pediatr Adolesc Health Care 2015;45(4):97-111. [http://dx.doi.org/10/1016/j.cppeds.2015.02.005] [ Links ]
11. Veugelers M, de Cat B, Muyldermans S, Reekmans G, Delande N, Frints S. Mutational analysis of the GPC3/GPC4 glypican gene cluster on Xq26 in patients with Simpson-Golabi-Behmel syndrome: Identification of loss-of-function mutations in the GPC3 gene. Hum Mol Genet 2000;9(9):1321-1328. [http://dx.doi.org/10.1093/hmg/9.9.1321] [ Links ]
12. Waterson J, Stockley T, Segal S, Golabi M. Novel duplication in glypican-4 as an apparent cause of Simpson-Golabi-Behmel syndrome. Am J Med Genet 2010;152A(12):3179-3181. [http://dx.doi.org/10.1002/ajmg.a.33450] [ Links ]
13. Sakazume S, Okamoto N, Yamamoto T, Kurosawa K, Numabe H, Ohashi Y. GPC3 mutations in seven patients with Simpson-Golabi-Behmel syndrome. Am J Med Genet 2007;143A(15):1703-1707. [http://dx.doi.org/10.1002/ajmg.a.31822] [ Links ]
14. Fokkema IF, Taschner PE, Schaafsma GC, Celli J, Laros JF, den Dunnen JT. LOVD v.2.0: The next generation in gene variant databases. Hum Mutat 2011;32(5):557-563. [http://dx.doi.org/10.1002/humu.21438] [ Links ]
15. National Center for Biotechnology Information. http://www.ncbi.nlm.nih.gov/gene/2719 (accessed 10 September 2015). [ Links ]
 Correspondence:
Correspondence:
C Spencer
careni.spencer@nhls.ac.za
Accepted 13 October 2015.

