










Serviços Personalizados
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkSAMJ: South African Medical Journal
versão On-line ISSN 2078-5135
versão impressa ISSN 0256-9574
SAMJ, S. Afr. med. j. vol.106 no.3 Pretoria Mar. 2016
http://dx.doi.org/10.7196/samj.2016.v106i3.10134
IN PRACTICE
DIAGNOSIS
Digitotalar dysmorphism: Molecular elucidation
A A VorsterI; P BeightonII; R S RamesarIII
ISenior Technical Officer in the MRC Human Genetics Research Unit in the Division of Human Genetics, Institute for Infectious Diseases and Molecular Medicine, Department of Pathology, Faculty of Health Sciences, University of Cape Town, South Africa, with an interest in molecular genetics of connective tissue disorders
IIAgeing medical geneticist in the Division of Human Genetics with a long-term interest in heritable disorders of the skeleton and connective tissue
IIIProfessor and Head of the Division of Human Genetics and Director of the MRC Human Genetics Research Unit
ABSTRACT
Dominantly inherited digitotalar dysmorphism (DTD), which is characterised by flexion contractures of digits and 'rocker-bottom' feet due to a vertical talus, was first described in a South African family of European stock in 1972. We review the clinical manifestations and document the molecular basis for DTD in this prototype family. This family was restudied in 1995 and 2006 and biological specimens were obtained for molecular studies. Since the distal arthrogryposes (DAs) are genetically heterogeneous, an unbiased approach to mutation identification was undertaken through whole-exome next-generation sequencing of DNA from a single DTD-affected female. Venous blood specimens were obtained for DNA banking and subsequent molecular studies. Analysis of the nine genes that had previously been shown to cause DAs revealed a pathogenic mutation in exon nine of TNNT3. The presence of the p.(Arg63His) missense mutation at position 63 of TNNT3 was confirmed through direct cycle sequencing of genomic DNA in six affected family members for whom DNA had been archived.
Background
Digitotalar dysmorphism (DTD; OMIM #126050) is an autosomal dominant disorder characterised by congenital fixed flexion deformities of the digits and 'rocker-bottom' feet due to a vertical position of the talus. The condition was originally delineated more than 40 years ago in five generations of a large South African (SA) kindred of European stock.[1]
The distal arthrogryposes (DAs) are a group of genetic disorders in which congenital contractures in the hands and feet are the main manifestations.[2] The individual members of the DA category have numerical designations 1 - 10, with subtypes indicated by letters in some instances. A few well-known eponymous genetic disorders with contractures plus craniofacial involvement have also been listed as DAs.
With ongoing elucidation of the molecular basis of the DAs, several genes that encode the contractile apparatus of the fast-twitch myofibrils have been implicated.[3] It has also emerged that there is considerable molecular heterogeneity in the DAs and that some conditions regarded as having autonomous syndromic status have identical mutational determinants.[4,5]
The prototype DTD family was restudied in 1995 and 2006, and biological specimens were obtained for molecular studies. A causative mutation in the TNNT3 (OMIM #600692) gene has now been identified.
The affected family
The proposita of the DTD family was born in SA in 1916 and initially examined in 1971 at the age of 55 years. At that time, nine affected persons in three generations were available for documentation.
Family photographs revealed that deceased individuals in two preceding generations had the disorder. The pedigree data were in keeping with autosomal dominant inheritance, with some variability in phenotypic expression and complete penetrance. The progenitor of the kindred had emigrated from Holland to SA during the 18th century, and there could be additional affected persons in the extended family in Europe.
The kindred was restudied in 1995 and in 2006, by which time three affected children in a new generation had been born and 11 affected persons in four generations were alive. Biological specimens for molecular investigations were obtained.
Clinical manifestations
The clinical manifestations in nine affected family members have been documented and depicted in the orthopaedic literature.[1] The main syndromic components are rigidities of the digits in the hands and feet together with vertical alignment of the talus bones in the ankles. Involvement of the extremities is usually bilateral and not necessarily symmetrical or of equal severity.
Hands
Flexion contracture of the digits, with some ulnar deviation, are consistent features. Severity and distribution are variable, but the deformities are fixed and neither progress nor ameliorate. The thumbs may lie in opposition, internal rotation movements being limited by a fleshy web. The soft tissues of the middle phalanges are attenuated, producing an appearance of digital narrowing and tightness of the overlying skin. The ventral surfaces of the fingers and palms are smooth with few markings, and a single palmar crease may be present. There is no evidence of vascular or neurological deficit, and the degree of physical handicap is usually mild. Radiological examination shows that the bones of the hands are entirely normal.
Feet
In the feet, vertical angulation of the talus bone is an important diagnostic manifestation which was present in 10 of the 12 family members for whom clinical information was available. This malpositioning produces the well-known 'rocker-bottom' convex configuration of the soles. The forefeet may be wide and flat with medial bulging of the insteps and flexion of the toes. As with the hands, the severity of involvement of the feet is variable. At the severe end of the spectrum, the fitting of shoes may be difficult and operative intervention may be required to facilitate ambulation.
Other manifestations
General health and intellectual function are normal, as are the facies, teeth, hair, nails, viscera and other bodily systems. Stature is normal for the generation and population group of the affected persons. The range of movements in the wrist, ankles and knees is limited to some extent in a minority of affected family members. In recent years two affected women have had caesarean sections for childbirth; the obstetric indications for these procedures are not known. Two family members have had bilateral carpal tunnel release operations in the wrists.
Methods
The family was reviewed in 1995 and again in 2006 when members of the new generation were examined and venous blood specimens were obtained for DNA banking and subsequent molecular studies. The specimens were collected with full signed consent, in accordance with established procedure.
Genomic DNA (gDNA) was extracted from peripheral blood lymphocytes using the salting-out method[6] and archived for 11 family members, of whom six had the classic DTD phenotype. For the identification of the disease-causing variant, whole-exome sequencing was performed for one patient (DTD1.3ISA) on the Illumina HiSeq 2000 (Illumina Inc., USA) using the NimbleGen V2 exome kit (Roche NimbleGen Inc., USA) for capture. A mean coverage of 34.5 times was achieved across all targeted bases to enable whole-exome variant calling. The variants identified in DTD1.3ISA were filtered for known pathogenic mutations from the ClinVar database[7,8] in nine genes (TPM2, MYBPC1, MYH3, TNNT3, TNNI2, PIEZO2, ECEL1, MYH8 and FBN2) that had previously been shown to be mutated in persons with DA (OMIM #108120).
To validate the pathogenic variant identified through whole-exome sequencing analysis of DTD1.3ISA, 100 ng of gDNA was amplified for exon nine of TNNT3 (RefSeq NM_006757.3) using 0.4 μΜ each of the sense (5'-CATGGCAATGTGGAGAAG-3') and antisense (5'-CTCTGATGAGGCAGCAGT-3') primers in a final polymerase chain reaction (PCR) volume of 25 μl containing 0.5 U GoTaq DNA polymerase, 1 χ colourless reaction buffer (Promega, USA) and 0.2 mM of each deoxynucleoside triphosphate (Bioline, UK). Following 5 minutes of denaturation at 95°C on the BioRad T100 thermal cycler (Bio-Rad, USA), DNA was amplified in 35 cycles of denaturation at 94°C for 30 seconds, with primer annealing at 60°C for 30 seconds and product extension at 72°C for 40 seconds. The reaction was concluded at 72°C for 7 minutes to allow for complete product extension. Direct cycle sequencing of the resulting fragments was completed using 0.08 μΜ of primers described for PCR with the BigDye direct cycle sequencing chemistry (Life Technologies, USA) on the 3130xl Genetic Analyser (Applied Biosystems, USA). DNA sequences of research participants were aligned to the reference TNNT3 (NG_013085.1) sequence using Unipro UGENE v1.14.2.[9]
This investigation received ethical approval from the UCT Faculty of Health Sciences Human Research Ethics Committee (REC REF 917/2014).
Results
Following whole-exome sequencing analysis of a person with DTD, 102 nucleotide variants were identified within the nine known genes that cause DAs. Twenty-one of these variants were described in the ClinVar database, and only one had a pathogenic designation (RCV000009468). The pathogenic variant, identified in TNNT3 (NM_006757.3), is a transition mutation at position 188 of the consensus coding sequence and is predicted to substitute an arginine for a histidine residue at amino acid position 63.
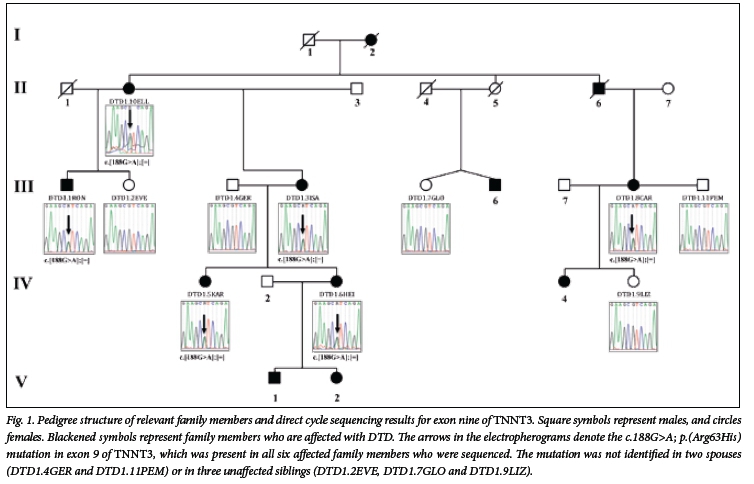
The presence of the c.188G>A mutation in the gDNA of DTD1.3ISA was confirmed through direct cycle sequencing of exon 9 of TNNT3 (Fig. 1, DTD1.3ISA). Following direct cycle sequencing of exon nine of TNNT3 in 10 extended-family members for whom DNA was archived, all affected individuals (n=6) were shown to carry the variant allele, while neither of the spouses (n=2) and none of the unaffected members (n=3) inherited the c.188G>A mutation.
Discussion
The prototype SA family with DTD is regarded as having a form of DA, and the vertical talus bones that produce the 'rocker-bottom' feet are a prime syndromic component.[1l In addition to DTD, a congenital vertical talus can occur sporadically and as a non-syndromic autosomal dominant trait.[10,11]
The issue of syndromic identity arose when a form of DTD associated with craniofacial abnormalities was documented.[12] In a series of 47 families with either DA2A or DA2B, Sung et al.[13] identified the TNNT3 c.188G>A; p.(Arg63His) mutation in a mother and her two DA2B-affected children. Specific phenotypic description was not provided for the family. The Sheldon-Hall type of DA also manifests significant facial features and a vertical talus; considerable non-allelic heterogeneity was demonstrated in this condition, including mutations in the gene TNNT3.[3]Thereafter, in 2011, Zhao et al.[14] documented five members of three generations of a Chinese family with the Sheldon-Hall phenotype and demonstrated that they had a specific mutation in TNNT3 (c.187C>T; p.R63C). These authors suggested that bilateral vertical talus or clubfoot might be a special characteristic of this hot-spot mutation. The TNNT3 mutation, reported by Sung et al.[13]in 2003, has now been identified in the prototype DTD family in SA. It is relevant that the craniofacial structures are entirely normal in this SA kindred, and the reason for the disparity in the genotype-phenotype correlation is unknown. Nevertheless, the recognition of a vertical talus as an indicator of a specific mutation is a useful concept.[13]
From the clinical standpoint, the detection of the specific TNNT3 mutation in the SA DTD family will facilitate genetic management. These measures could include diagnostic confirmation, antenatal diagnosis and genetic counselling, among others. In the future, the elucidation of the role of the causative TNNT3 mutation in the pathogenesis of DTD could have implications for targeted forms of therapy.
Acknowledgements. Support for the project from the South African Medical Research Council and the National Research Foundation is acknowledged with gratitude by PB. We thank Dr Μ Solomons from the Life Vincent Pallotti Hospital for assistance in obtaining DNA from DTD patients for molecular investigations.
References
1. Sallis JG, Beighton P. Dominantly inherited digito-talar dysmorphism. J Bone Joint Surg Br 1972;54(3):509-515. [ Links ]
2. Hall JG, Reed SD, Greene G. The distal arthrogryposes: Delineation of new entities - review and nosologic discussion. Am J Med Genet 1982;11(2):185-239. [http://dx.doi.org/10.1002/ajmg.1320110208] [ Links ]
3. Toydemir RM, Bamshad MJ. Sheldon-Hall syndrome. Orphanet J Rare Dis 2009;4:11. [http://dx.doi.org/10.1186/1750-1172-4-11] [ Links ]
4. Beck AE, McMillin MJ, Gildersleeve HI, et al. Spectrum of mutations that cause distal arthrogryposis types 1 and 2B. Am J Med Genet A 2013;161A(3):550-555. [http://dx.doi.org/10.1002/ajmg.a.35809] [ Links ]
5. McMillin MJ, Beck AE, Chong JX, et al. Mutations in PIEZO2 cause Gordon syndrome, Marden-Walker syndrome, and distal arthrogryposis type 5. Am J Hum Genet 2014;1(5):734-744. [http://dx.doi.org/10.1016/j.ajhg.2014.03.015] [ Links ]
6. Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res 1988;16(3):1215. [http://dx.doi.org/10.1093/nar/16.3.1215] [ Links ]
7. Landrum MJ, Lee JM, Riley GR, et al. ClinVar: Public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res 2014;42(1):D980-D985. [http://dx.doi.org/10.1093/nar/gkt1113] [ Links ]
8. ClinVar. http://www.ncbi.nlm.nih.gov/clinvar/ (accessed 7 September 2014). [ Links ]
9. Okonechnikov K, Golosova O, Fursov M. Unipro UGENE: A unified bioinformatics toolkit. Bioinformatics 2012;28(8):1166-1167. [http://dx.doi.org/10.1093/bioinformatics/bts091] [ Links ]
10. Hamanishi C. Congenital vertical talus: Classification with 69 cases and new measurement system. J Pediatr Orthop 1984;4(3):318-326. [http://dx.doi.org/10.1097/01241398-198405000-00007] [ Links ]
11. Stern HJ, Clark RD, Stroberg AJ, Shohat M. Autosomal dominant transmission of isolated congenital vertical talus. Clin Genet 1989;36(6):427-430. [ Links ]
12. Kantaputra PN, Chalidapong P, Visrutaratna P. Digitotalar dysmorphism with craniofacial and other new associated abnormalities. Clin Dysmorphol 2001;10(3):171-175. [http://dx.doi.org/10.1097/00019605-200107000-00003] [ Links ]
13. Sung SS, Brassington AM, Krakowiak PA, Carey JC, Jorde LB, Bamshad M. Mutations in TNNT3 cause multiple congenital contractures: A second locus for distal arthrogryposis type 2B. Am J Hum Genet 2003;73(1):212-214. [http://dx.doi.org/10.1086/376418] [ Links ]
14. Zhao N, Jiang M, Han W, et al. A novel mutation in TNNT3 associated with Sheldon-Hall syndrome in a Chinese family with vertical talus. Eur J Med Genet 2011;54(3):351-353 [http://dx.doi.org/10.1016/j.ejmg.2011.03.002] [ Links ]
 Correspondence:
Correspondence:
A A Vorster
anna.vorster@uct.ac.za
Accepted 14 October 2015.


{kind=link}