










Serviços Personalizados
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkSAMJ: South African Medical Journal
versão On-line ISSN 2078-5135
versão impressa ISSN 0256-9574
SAMJ, S. Afr. med. j. vol.106 no.3 Pretoria Mar. 2016
http://dx.doi.org/10.7196/samj.2016.v106i3.10170
EDITORIAL
Could we offer mitochondrial donation or similar assisted reproductive technology to South African patients with mitochondrial DNA disease?
Mitochondrial disease comprises a highly heterogeneous group of disorders in which mitochondrial function is impaired as a result of mutations located either in mitochondrial DNA (mtDNA) itself or in a large number of nuclear genes involved in mitochondrial function.[1] In 2015 the House of Commons in the UK voted to endorse the use of pioneering in vitro fertilisation (IVF) techniques to protect future generations from the risk of mtDNA disease by the use of mitochondrial donation.
More than one method to implement mitochondrial donation has been suggested in the literature. Options include the transfer of pronuclei from the parents into a mitochondrial donor zygote,[2] or alternatively metaphase II spindle transfer into a mitochondrial donor oocyte.[3] In both these scenarios, the offspring inherit their nuclear DNA (nDNA) from the gametes contributed by both parents but their mtDNA from another female (donor), introducing the three-parent scenario that has sparked controversy and debate.[4,5]
Mitochondrial DNA has several properties that distinguish it from nDNA: it is strictly maternally inherited and does not undergo recombination; it is not wrapped around histone proteins for protection, and is consequently exposed to high levels of mutation-generating free radicals; and there are many copies per cell, ranging from two up to several thousand. In addition, the mutation rate in mtDNA is about 10 times greater than in nDNA, and replication errors are less likely to be repaired. The high mutation rate coupled with the polyploid nature of the mitochondrial genome gives rise to an important feature of mitochondrial genetics, namely homoplasmy and heteroplasmy. In simple terms, homoplasmy is when all copies of the mitochondrial genome in a given cell/tissue/organ are identical, and heteroplasmy is when there is a mixture of two or more mitochondrial genotypes.[1] Mutations in mtDNA often lead to deleterious consequences. Some mutations affect all copies of the mitochondrial genome (homoplasmic mutation), whereas others are only present in some copies of the mitochondrial genome (heteroplasmic mutation). In the presence of heteroplasmy, there is a mutation threshold level that is important both for the clinical expression of the disease and for biochemical defects. The precise threshold depends on the pathogenicity and the tissue involved, resulting in a complex and heterogeneous pattern of disease presentation.[1] Another level of complexity is introduced when a population-specific mutation is associated with the aetiology and presentation of a disease.[6,7] Differences in disease presentation are also found between adult and paediatric patients: adults may present with one of the characteristic mtDNA phenotypes such as mitochondrial encephalomyopathy, lactic acidosis and stroke-like episodes (MELAS), whereas paediatric patients tend to present early with severe, complex neurological manifestations, hepatopathy, renal tubulopathy, endocrinopathy or cardiomyopathy with rapid progression.
The current regulations in the UK state that mitochondrial IVF techniques should be limited to selected cases where 'there is a particular risk that the patient's egg or an embryo created using the patient's egg will carry a mtDNA abnormality and that there is a significant risk that a child born from the use of that egg will have or develop a serious mitochondrial disease'.[8] Suitable infrastructure and capacity to perform the necessary molecular diagnosis together with interpretation of clinical data linking a particular mtDNA mutation to the disease is required. While many laboratories in the UK are capable of providing such services, it is timely for us to examine the level of service capability in South Africa (SA).
Although individually rare, as a group mitochondrial genetic disorders are thought to be responsible for a substantial proportion of inherited metabolic disease. An estimated prevalence of 1:5 000 -1:10 000 has been reported in several (mostly developed-nation) populations.[9-13] However, the prevalence of mitochondrial genetic disorders is still largely unknown in SA. To stimulate dialogue on the status of mitochondrial disease diagnosis and research in SA, a workshop was convened in Potchefstroom, North West Province, in November 2014 that enabled a small group of local scientists and clinicians to engage with colleagues from the UK on this subject.[7]
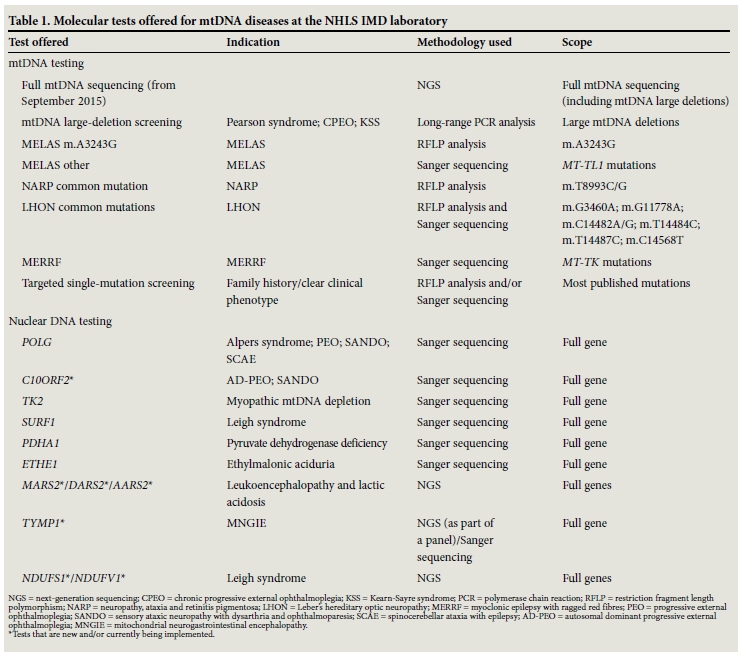
Two centres are involved in mtDNA diagnostics in SA: the Inherited Metabolic Diseases (IMD) Laboratory of the National Health Laboratory Service (NHLS) in Cape Town and the Mitochondria Research Laboratory in the Centre for Human Metabolomics (CHM) at NorthWest University (NWU) in Potchefstroom. The IMD laboratory serves as the referral centre for mtDNA disease genetic studies. Molecular testing for deletions and common point mutations associated with classic phenotypes such as MELAS is offered, as well as limited targeted nuclear gene sequencing (Table 1). Referrals for common mutations were only positive in about 6% of >1 000 cases examined between 1990 and 2012.[7] This low percentage can be attributed in part to incomplete clinical information received with specimens sent for molecular testing, lack of capacity to track and test maternal relatives, and inappropriate requests. Often it is difficult to establish whether: (i) the correct tissue was supplied; (ii) the test requested matched the phenotype; or (iii) other possible diseases were excluded. From August 2015, this laboratory has shifted its diagnostic strategy to that of sequencing of the entire mtDNA genome using a next-generation sequencing approach, as well as widening its nuclear gene sequencing repertoire for non-mtDNA inherited mitochondrial disorders (Table 1). Clinical screening is being strengthened to try to improve diagnostic efficiency.
The Mitochondria Research Laboratory in the CHM at NWU, which is not affiliated to the NHLS IMD Laboratory, has since 1998 approached mtDNA disease investigations mainly in cohort investigations with the Department of Paediatrics, University of Pretoria, as well as ad hoc cases from elsewhere in SA. The laboratory approaches molecular investigations using full-length mtDNA next-generation sequencing, which follows respiratory-chain single-enzyme kinetics in muscle biopsies and urine metabolomics investigations from clinically selected patients.[14,15] A collaborative research project undertaken by this laboratory with the University of Pretoria involved a cohort study of >200 well-characterised paediatric patients with clinically diagnosed mitochondrial disease drawn from Limpopo, Gauteng and Mpumulanga provinces. Respirometry and single-enzyme analyses were conducted in muscle biopsies to confirm mitochondrial disease in these patients. Even at this high level of resolution, <1% of mtDNA mutations detected were linked with known pathogenic mtDNA mutations, indicating that many novel mtDNA mutations with as yet unresolved impact were present in this cohort.[6]
In order to enter the debate on whether or not SA should offer prenatal testing or interventions such as the IVF technique described above to patients with mtDNA disease, we should at least be able to predict the number of women who would benefit in this country, as was done for the UK and the USA.[4] Using the data presented above and considering that there are <70 genetically confirmed cases in a country with a population of ~56 million (i.e. 1:900 000 compared with an estimated 1:5 000 worldwide), it would appear that the number of undiagnosed cases in SA is staggeringly high.
Furthermore, from observations made on other inherited metabolic disorders, such as type 1 glutaric aciduria, galactosaemia and cystinosis,[16-18] it is becoming increasingly evident that the mutations commonly reported do not appear to reflect the disease-causing mutations among black African populations.[7] More research is required locally to understand patterns of mtDNA variation in black African groups so that a judicious assessment can be made when attempting to distinguish those mutations that are associated with mtDNA disease from those that have evolved stochastically over time. Given the gaps that exist in identifying mtDNA and nDNA mutations linked with mtDNA disease, coupled with the lack of adequate patient biochemical and clinical information, the situation in SA is far from optimal for diagnosing mtDNA disease.
What should be done to improve the situation? A more integrated approach involving the availability of clinical, biochemical, molecular and imaging data, as illustrated in Fig. 1, is required to refine the diagnosis of mtDNA disease. Clinicians play a vital role, as robust clinical assessment and diagnosis is required prior to submitting samples for molecular screening. Often the pattern of mtDNA disease symptoms only becomes apparent over time with consistent follow-up, usually by the same clinician. SAs overloaded primary healthcare system is not designed to focus on rare disorders, and the time required to conduct proper examinations for more complicated cases is often not available. However, symptoms such as growth failure, deafness, epilepsy and muscle weakness are easily recognisable and are referable.
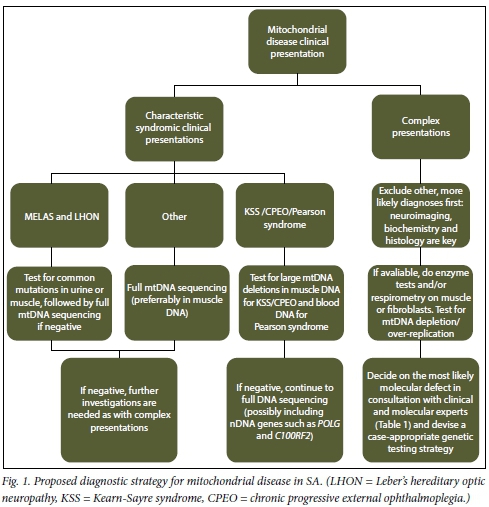
Moreover, good support in terms of access to neuroimaging (magnetic resonance imaging) and experienced histopathology services are also necessary. For the diagnosis of mitochondrial disease, enzyme analyses in tissue biopsies along with the more extensive biochemical analyses (e.g. respirometry and other functional and structural investigations) are needed to support molecular genetic investigations. The availability of next-generation sequencing platforms makes sequencing of the entire mitochondrial genome feasible and cost-effective for consideration as a routine diagnostic test. This will assist in determining at least the involvement of the mitochondrial genome when suggested from clinical observations, particularly in adult patients. However, strong collaborations between diagnostic and research teams are key in linking pathogenicity to novel or known mtDNA mutations detected with this technology.[19,20]
Mitochondrial disease is present in all SA populations, but the majority of patients have not been able to obtain a molecular diagnosis. The declining cost of conducting whole-mtDNA sequencing will allow identification of mtDNA mutations and improve our capacity for accurate diagnosis and appropriate patient counselling and management. Extension of molecular testing to look at nuclear genes should enhance this capacity further. Both should be guided by supportive biochemical testing. Now that these strategies are being put in place to offer a basic but robust service for diagnosis of mtDNA diseases, it is time for SA to initiate a debate on the IVF technique that has been topical elsewhere.
Surita Meldau
Division of Chemical Pathology, Faculty of Health Sciences, University of Cape Town, South Africa, and National Health Laboratory Service, Cape Town
Gillian Riordan
Department of Paediatrics and Child Health, Faculty of Health Sciences, University of Cape Town, South Africa, and Red Cross War Memorial Children's Hospital, Cape Town
Francois van der Westhuizen
Human Metabolomics, North-West University, Potchefstroom, South Africa
Joanna L Elson
Mitochondrial Research Group, Institute of Genetic Medicine, Newcastle University, Newcastle-upon-Tyne, UK, and Human Metabolomics, North-West University, Potchefstroom, South Africa
Izelle Smuts
Department of Paediatrics and Child Health, Faculty of Health Sciences, University of Pretoria, South Africa, and Steve Biko Academic Hospital, Pretoria
Michael S Pepper
Department of Immunology and Institute for Cellular and Molecular Medicine, Faculty of Health Sciences, University of Pretoria, South Africa
Himla Soodyall
Division of Human Genetics, Faculty of Health Sciences, University of the Witwatersrand, Johannesburg, South Africa, and National Health Laboratory Service, Johannesburg
References
1. Tuppen HA, Blakely EL, Turnbull DM, Taylor RW. Mitochondrial DNA mutations and human disease. Biochim Biophys Acta 2010;1797(2):113-128. [http://dx.doi.org/10.1016/j.bbabio.2009.09.005] [ Links ]
2. Craven L, Tuppen HA, Greggains GD, et al. Pronuclear transfer in human embryos to prevent transmission of mitochondrial DNA disease. Nature 2010;465(7294):82-85. [http://dx.doi.org/10.1038/nature08958] [ Links ]
3. Tachibana M, Sparman M, Sritanaudomchai H, et al. Mito-chondrial gene replacement in primate offspring and embryonic stem cells. Nature 2009;461(7262):367-372. [http://dx.doi.org/10.1038/nature08368] [ Links ]
4. Gorman GS, Grady JP, Ng Y, et al. Mitochondrial donation -how many women could benefit? N Engl J Med 2015;372(9):885-887. [http://dx.doi.org/10.1056/NEJMc1500960] [ Links ]
5. Reznichenko AS, Huyser C, Pepper MS. Mitochondrial transfer: Ethical, legal and social implications in assisted reproduction. S Afr J Bioethics Law 2015;8(2 Suppl 1):32-35. [ Links ]
6. Van der Walt EM, Smuts I, Taylor RW, et al. Characterization of mtDNA variation in a cohort of South African paediatric patients with mitochondrial disease. Eur J Hum Genet 2012;20(6):650-656. [http://dx.doi.org/10.1038/ejhg.2011.262] [ Links ]
7. Van der Westhuizen FH, Sinxadi PZ, Dandara C, et al. Understanding the implications of mitochondrial DNA variation in the health of black southern African populations: The 2014 Workshop. Hum Mutat 2015;36(5):569-571. [http://dx.doi.org/10.1002/humu.22789] [ Links ]
8. Barber DS, Border DP. Mitochondrial Donation. UK House of Commons Library. http://researchbriefings.files.parliament.uk/documents/SN06833/SN06833.pdf2015 (accessed 29 July 2015). [ Links ]
9. Schaefer AM, McFarland R, Blakely EL, et al. Prevalenceof mitochondrial DNA disease in adults. Ann Neurol 2008;63(1):35-39. [http://dx.doi.org/10.1002/ana.21217] [ Links ]
10. Gorman GS, Schaefer AM, Ng Y, et al. Prevalence of nuclear and mitochondrial DNA mutations related to adult mitochondrial disease. Ann Neurol 2015;77(5):753-759. [http://dx.doi.org/10.1002/ana.24362] [ Links ]
11. Skladal D, Halliday J, Thorburn DR. Minimum birth prevalence of mitochondrial respiratory chain disorders in children. Brain 2003;126(8):1905-1912. [http://dx.doi.org/10.1093/brain/awg170] [ Links ]
12. Darin N, Oldfors A, Moslemi AR, Holme E, Tulinius M. The incidence of mitochondrial encephalomyopathies in childhood: Clinical features and morphological, biochemical, and DNA anbormalities. Ann Neurol 2001;49(3):377-383. [http://dx.doi.org/10.1002/ana.75] [ Links ]
13. Applegarth DA, Toone JR, Lowry RB. Incidence of inborn errors of metabolism in British Columbia, 1969-1996. Pediatrics 2000;105(1):e10. [http://dx.doi.org/10.1542/peds.105.1.e10] [ Links ]
14. Smuts I, Louw R, du Toit H, Klopper B, Mienie LJ, van der Westhuizen FH. An overview of a cohort of South African patients with mitochondrial disorders. J Inherit Metab Dis 2010;33(Suppl 3):S95-S104. [http://dx.doi.org/10.1007/s10545-009-9031-8] [ Links ]
15. Venter L, Lindeque Z, Jansen van Rensburg P, van der Westhuizen F, Smuts I, Louw R. Untargeted urine metabolomics reveals a biosignature for muscle respiratory chain deficiencies. Metabolomics 2015;11(1):111-121. [http://dx.doi.org/10.1007/s11306-014-0675-5] [ Links ]
16. Owen EP, Nandhlal J, Leisegang F, van der Watt G, Nourse P, Gajjar P. Common mutation causes cystinosis in the majority of black South African patients. Pediatr Nephrol 2015;30(4):595-601. [http://dx.doi.org/10.1007/s00467-014-2980-7] [ Links ]
17. Van der Watt G, Owen EP, Berman P, et al. Glutaric aciduria type 1 in South Africa - high incidence of glutaryl-CoA dehydrogenase deficiency in black South Africans. Mol Genet Metab 2010;101(2-3):178-182. [http://dx.doi.org/10.1016/j.ymgme.2010.07.018] [ Links ]
18. Henderson H, Leisegang F, Brown R, Eley B. The clinical and molecular spectrum of galactosemia in patients from the Cape Town region of South Africa. BMC Pediatr 2002;2:7. [http://dx.doi.org/10.1186/1471-2431-2-7] [ Links ]
19. Yarham JW, Al-Dosary M, Blakely, EL, et al. A comparative analysis approach to determining the pathogenicity of mitochondrial tRNA mutations. Hum Mutat 2011;32(11):1319-1325. [http://dx.doi.org/10.1002/humu.21575] [ Links ]
20. Montoya J, Lopez-Gallardo E, Herrero-Martin MD, et al. Diseases of the human mitochondrial oxidative phosphorylation system. In: Espinós C, Felipo V, Palau F, eds. Inherited Neuromuscular Diseases: Translation from Pathomechanisms to Therapies. Dordrecht, Netherlands: Springer, 2009:47-67. [ Links ]
 Correspondence:
Correspondence:
S Meldau
surita.meldau@nhls.ac.za


{kind=link}