










Servicios Personalizados
Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkSAMJ: South African Medical Journal
versión On-line ISSN 2078-5135
versión impresa ISSN 0256-9574
SAMJ, S. Afr. med. j. vol.105 no.12 Pretoria dic. 2015
http://dx.doi.org/10.7196/samj.2015.v105i12.10226
CONTINUING MEDICAL EDUCATION
ARTICLE
Meeting the challenges in the diagnosis of inflammatory myopathies
M Manie
MB ChB, FCP (SA), MMed, Cert Rheumatology (SA). Division of Rheumatology, Department of Medicine, Faculty of Medicine and Health Sciences, Stellenbosch University, Cape Town, South Africa
ABSTRACT
Inflammatory myopathy (IM) is a rubric term to describe a heterogeneous group of muscle diseases typified by dermatomyositis and polymyositis. The current classifications are unsatisfactory, but IM associated with other connective tissue diseases (CTDs), such as systemic lupus erythematosus, underlying malignancy and HIV, should also be included. Although uncommon, IM should always be considered in a patient who presents with proximal weakness of gradual onset and has raised serum muscle enzymes.
The diagnosis may be obvious if the patient has diagnostic skin signs such as heliotropic rash (peri-orbital discoloration) and Gottron's lesions (typically on the extensor surfaces of the fingers). In the absence of obvious skin manifestations, other features of a CTD such as Raynaud's phenomenon, abnormal capilloroscopy and the presence of serum antinuclear factor antibody should be searched for.
Conditions that mimic IM include other causes of myopathy such as endocrine disorders, adverse effects of medication, metabolic myopathies and muscle dystrophies. Atypical features suggesting an alternative diagnosis are acute onset, severe pain, assymmetrical involvement, distal weakness and wasting.
Appropriate investigations include a chest radiograph indicating interstitial lung disease or malignancy. Electromyography and muscle biopsy are useful in cases where other diagnoses are suspected.
Inflammatory myopathy (IM) is an umbrella term for a group of muscle diseases exemplified by dermatomyositis (DM) and polymyositis (PM). Collectively, these conditions were considered to be rare, but seem to be more common than previously thought. Making an incorrect diagnosis may be costly, as delays in initiating treatment may result in morbidity and mortality. As a disease entity IM is generally poorly understood compared with other connective tissue diseases (CTDs).[1,2]
Diagnosis
The Bohan and Peter criteria[3,4] for diagnosing IM are listed below. Initially published in 1975, these are still commonly referred to but have limited usefulness in clinical practice:
-
Symmetric proximal muscle weakness.
-
Elevated serum muscle enzymes.
-
Myopathic changes on electromyography(EMG).
-
Typical cutaneous manifestations of DM (the clinical feature distinguishing DM from PM).
-
Characteristic muscle biopsy abnormalities and the absence of histopathological signs of other muscle diseases.
Classification
Current classifications of IM are fraught with problems.[5] From a rheumatologist's point of view it may be prudent to view IMs as being associated with a CTD or having a definite auto-immune aetiology. Therefore, inclusion body myositis (IBM) would probably not find a place in this sub-classification. A useful classification of IM is the following:
1. DM.
2. PM.
3. DM and PM overlapping with a CTD, such as systemic lupus erythematosus (SLE)/scleroderma.
4. Points 1 - 3 have another cause, such as malignancy or HIV.
5. 'Undifferentiated' IM, such as necrotising autoimmune inflammatory myopathy (NAM).
1. Dermatomyositis
The diagnosis of DM is generally based on the features of proximal muscle weakness of gradual onset, raised muscle enzymes, e.g. creatinine kinase (CK), and cutaneous features. The skin manifestations of DM may range from highly typical to subtle.[6]
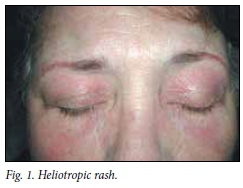
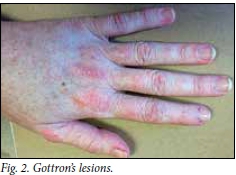
Pathognomonic features are the heliotropic rash and Gottron's lesions. The heliotropic rash (Fig. 1) is characterised by peri-orbital swelling and violaceous discoloration. This may be very severe and conspicuous, or subtle and transient, and may have disappeared when the patient presents with weakness. Gottron's lesions (Fig. 2) are violaceous papules or scaly plaques on the extensor surfaces of the fingers, e.g. the knuckles.
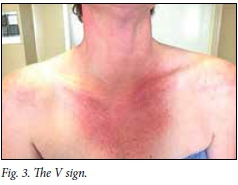
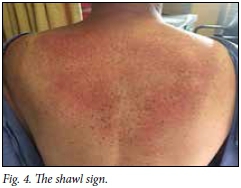
Very suspicious features include well-defined areas of erythematous swelling in a typical distribution, resulting in the V (Fig. 3) and shawl (Fig. 4) signs.
Less specific, but not uncommon skin manifestations include poikiloderma, seborrhoeic dermatitis/psoriasiform changes of the scalp and diffuse erythroderma.
Amyopathic DM is a variant of DM that spares muscle and does not present with weakness, but may manifest with weakness in the course of the disease. The diagnosis is made on the findings of diagnostic cutaneous signs, such as the heliotropic rash referred to above.
In cases where the cutaneous signs are not convincing, a skin biopsy may be performed. The finding of an interface dermatitis is fairly specific for DM but also occurs in SLE.
2. Polymyositis
PM lacks the skin characteristics of DM but is suspected if there are other features of a CTD.M These include Raynaud's phenomenon, peri-ungual swelling or erythema, 'raggedy' cuticles or other features of the antisynthetase syndrome. This syndrome refers to a patient with PM or DM with the typical findings of constitutional symptoms such as fever, polyarthritis, mechanic's hands (Fig. 5), interstitial lung disease and the presence of anti-Jo-1 antibody in the serum.
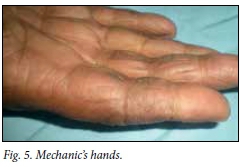
The diagnosis of PM is more challenging because of the paucity of cutaneous clues. PM is currently considered a rare cause of IM. Previously, the diagnosis was largely clinical and the condition was probably over-diagnosed. In retrospect, in many patients labelled as having PM, there were other causes for the proximal weakness, such as those mentioned in the differential diagnosis below.[7]
When taking a history, the astute physician should look for clues, such as Raynaud's phenomenon and inflammatory pain affecting the joints. On examination, one should look for other features of a CTD, such as abnormal capillaroscopy, puffy hands and interstitial lung disease. A dermatoscope, if available, is a preferred alternative to an ophthalmoscope to identify vascular abnormalities such as tortuosity and loss of capillaries in the nail bed. Occasionally, the diagnosis may lean heavily on the absence of another cause of the myopathy.
3. Inflammatory myopathy partially overlapping with another connective tissue disease
DM or PM may overlap with a CTD such as SLE, scleroderma or mixed CTD. However, deciding whether the IM is part of the underlying CTD and not necessarily overlapping with the IM may be difficult.
4. Inflammatory myopathy with underlying malignancy or HIV
It is well known that DM (up to 15%) and to a lesser extent PM are often associated with an underlying malignancy such as carcinoma of the breast or lung. IM associated with HIV is discussed below.
5. 'Undifferentiated inflammatory myopathy', e.g. necrotising auto-immune myopathy (discussed below)
Differential diagnosis
Reviews of the histopathology of patients clinically diagnosed with IM not infrequently result in an alternative diagnosis.'81 These include:
• Inclusion body myositis
Some clinicians, in particular neurologists, include IBM as a subtype of IM, but the underlying aetiopathogenesis is a degenerative muscle disorder with little underlying inflammation, akin to an Alzheimer's' of muscle. It is characterised by slow onset of action, characteristically over many years, and atypical features such as distal muscle weakness, asymmetrical involvement, muscle atrophy and a poor response to steroids. The condition was previously regarded as exceedingly rare, but may be more common as retrospective reviews have uncovered many cases originally diagnosed as PM. It is also a rare cause of myopathy in individuals with HIV.
• Endocrine/electrolyte disorders
It is important to exclude hyper- and hypothyroidism. The latter, apart from causing proximal weakness, may also cause elevation of the serum CK. In the realm of the 'metabolic' myopathies, it is important to rule out hypokalaemia, hypo- and hypercalcaemia, osteomalacia and Cushing's syndrome.
• Adverse effects of medication
Steroid-induced myopathy has to be considered in patients on maintenance oral steroids, e.g. those with poorly controlled asthma. Muscle weakness may occur in the absence of other features of Cushing's syndrome.
Although myalgia is a common symptom associated with the use of statins, weakness is rare. The mechanism is poorly understood but may be due to dose-related direct muscle toxicity and may very rarely be immune mediated'91 (discussed below).
Colchicine may cause the feared side-effect of a myoneuropathy, which may be irreversible. Patients particularly at risk of this rare complication are those with underlying renal impairment and the concomitant use of a macrolide antibiotic, e.g. clarithromycin.
Chloroquine is frequently used in rheumatic diseases such as rheumatoid arthritis and SLE, but fortunately the myopathy associated with its use is very rare.
Zidovudine is a well-known cause of HIV-associated muscle weakness owing to a mitochondrial myopathy.
• Diseases with pathology arising from the neuromuscular junction
Myasthenia gravis is a rare condition always worth considering, as the clues such as facial muscle weakness, ptosis and fatiguability may not always be obvious.
The Lambert-Eaton myasthenic syndrome is a disorder occurring in the setting of an underlying malignancy such as bronchus carcinoma.
• Muscle dystrophies
Limb girdle dystrophy. A family history and muscle atrophy involving the shoulder and pelvic girdle may be important clues.
Myotonic dystrophy. This is characterised by myotonia and additional findings of a family history, cataracts, cognitive impairment and impaired glucose tolerance.
• Metabolic myopathies
These conditions usually have a hereditary component and noteworthy features are episodes of acute myalgia with associated myoglobinuria and accompanying dark-coloured urine.
Atypical features that should suggest alternative diagnoses Severe muscle pain. Myalgia occurs in half of patients with an IM and is generally not a prominent feature. Severe pain suggests an infection or a metabolic cause. Pain mimics weakness, e.g. a typical patient with polymyalgia rheumatica may have shoulder girdle pain but present with 'weakness' rather than pain.
Acute onset. This should raise suspicion of an infective cause.
Normal or only slightly raised muscle enzymes. Although occasionally found in IM, this is unusual.
Constitutional symptoms such as fever and weight loss. These occur more frequently in patients whose IM partially overlaps with another CTD such as SLE. If present, this should also prompt a search for an underlying malignancy or HIV.
Wasting, asymmetrical weakness and disproportionate distal weakness. These suggest a neurological cause such as motor neuron disease or IBM.
Global areflexia. This should raise suspicion of chronic demye-linating polyneuropathy, which may uncommonly present with proximal rather than distal weakness.
Myopathies due to HIV and NAM
These diseases may cause IM, but are worthwhile highlighting as they may cause weakness via other mechanisms.
HIV may cause a clinical picture indistinguishable from PM clinically and histologically, usually occurs relatively early in the disease, and usually responds well to steroids. HIV may also cause muscle weakness on the basis of 'non-immune' mechanisms.[10-12] The nucleotide reverse transcriptase inhibitors may cause a mitochondrial myopathy with the diagnostic ragged red fibres on muscle histology. The chronic HIV wasting syndrome is the most common cause of muscle weakness in this illness and occurs in the late stages of the disease - it is usually not amenable to immunosuppressive therapy. IBM is a rare disease and may also occur in the setting of HIV. Myasthenia gravis and NAM (discussed below) are other rare illnesses linked to HIV.
NAM is a rare but important cause of myopathy.[13,14] It is associated with (i) CTDs such as SLE, scleroderma and mixed CTD; (ii) malignancy, such as carcinoma of the bronchus; (iii) statin use, although the myopathy caused by statins is generally via direct muscle toxicity and not immune mediated; and (iv) HIV or other viral infections. Knowledge about this condition is important as it requires treatment with immunosuppressive therapy such as that conventionally used for the treatment of IM. Anti-signal recognition particle antibody is a myositis-specific antibody associated with NAM, but is generally only used in research settings.
Appropriate investigations
With regard to the involvement of organ systems other than muscle and skin it is worthwhile trying to identify interstitial lung disease as it is reasonably common - in some studies affecting >50% of patients with IM. Cardiac involvement is rare.
Serological tests. The antinuclear factor test is useful as a screening test to detect underlying CTD. The anti-Jo-1 antibody test, although not sensitive, is relatively specific for interstitial lung disease and the antisynthetase syndrome mentioned above. Myositis-specific antibodies other than anti-Jo-1 have a very small role in current clinical practice.
Magnetic resonance imaging (MRI). MRI of muscle may help to distinguish between active inflammation and muscle necrosis, but its future role is unclear.
EMG. This is reserved for patients where there is doubt about the presence of active muscle necrosis, such as those with a normal or slightly raised CK.
Histopathology. Although there are characteristic differences in the pattern of the inflammatory infiltrate between DM and PM, a muscle biopsy for histology is not routine practice. A more compelling indication for muscle biopsy is to shed light on the diagnosis of patients who cannot be confidently diagnosed as having IM. A muscle biopsy is an invasive procedure and it may be prudent to subject likely cases to a trial of immunosuppressive therapy, with a satisfactory response to therapy supporting the diagnosis of IM. Advocates of improved access to muscle histopathology feel that this will result in more accurate diagnoses and better decisions with regard to therapy.[15]
Conclusion
IM, although uncommon, is an important cause of myopathy, which should always be considered in the differential diagnosis of a patient who presents with proximal weakness. The clinical context, particularly the possible presence of CTD or other systemic illnesses, is an important guide to the diagnosis.
Summary
-
IM should always be considered in a patient who presents with proximal weakness of gradual onset.
-
In the absence of obvious skin manifestations, other features of CTDs such as Raynaud's phenomenon, abnormal capilloroscopy and the presence of serum antinuclear factor antibody should be searched for.
-
Atypical features suggesting an alternative diagnosis are acute onset, severe pain, assymmetrical involvement, distal weakness, areflexia and wasting.
-
EMG is useful to distinguish neurological from myopathic causes.
-
Muscle biopsy is mandatory to distinguish between IM and other myopathies in the absence of obvious characteristic features.
Acknowledgements. I wish to thank Prof. Razeen Davids for proofreading the manuscript. Special thanks to the Division of Dermatology, Faculty of Medicine and Health Sciences, Stellenbosch University, for providing some of the figures.
References
1. Dalakas MC. Inflammatory muscle diseases. N Engl J Med 2015;372:1734-1747. [http://dx.doi.org/10.1056/NEJMra1402225] [ Links ]
2. Dalakas MC, Hohlfeld R. Polymyositis and dermatomyositis. Lancet 2003;362:971-982. [http://dx.doi.org/10.1016/S0140-6736(03)14368-1] [ Links ]
3. Bohan A, Peter JB. Polymyositis and dermatomyositis. N Engl J Med 1975;292:344-347. [http://dx.doi.org/10.1056/NEJM197502132920706] [ Links ]
4. Bohan A, Peter JB. Polymyositis and dermatomyositis. N Engl J Med 1975;292:403-407. [http://dx.doi.org/10.1056/NEJM197502202920807] [ Links ]
5. Christopher-Stine L. Neurologists are from Mars. Rheumatologists are from Venus: Differences in approach to classifying the idiopathic inflammatory myopathies. Curr Opin Rheumatol 2010;22:623-626. [http://dx.doi.org/10.1097/BOR.0b013e32833f8f72] [ Links ]
6. Wortmann RL. Myopathies. Rheum Dis Clin N Am 2011;37(2). [http://dx.doi.org/10.1016/j.rdc.2011.02.001] [ Links ]
7. Van der Meulen MFG, Bronner IM, Hoogendijk JE, et al. Polymyositis: An overdiagnosed entity. Neurology 2003;61:316-321. [http://dx.doi.org/10.1212/WNL.6L3.316] [ Links ]
8. Michelle EH, Mammen AL. Myositis mimics. Curr Rheumatol Rep 2015;17: 1-8. [http://dx.doi.org/10.1007/s11926-015-0541-0] [ Links ]
9. Mammen AL, Amato AA. Statin myopathy: A review of recent progress. Curr Opin Rheumatol 2010;22:644-650. [http://dx.doi.org/10.1097/BOR.0b013e32833f0fc7] [ Links ]
10. Heckmann JM, Pillay K, Hearn AP, Kenyon C. Polymyositis in African HIV-infected subjects. Neuromuscul Disord 2010;20:735-739. [http://dx.doi.org/10.1016/j.nmd.2010.06.007] [ Links ]
11. Nguyen BY, Reveille JD. Rheumatic manifestations associated with HIV in the highly active antiretroviral therapy era. Curr Opin Rheumatol 2009;21:404-410. [http://dx.doi.org/10.1097/BOR.0b013e32832c9d04] [ Links ]
12. Louthrenoo W. Rheumatic manifestations of human immunodeficiency virus infection. Curr Opin Rheumatol 2008;20:92-99. [http://dx.doi.org/10.1097/BOR.0b013e3282f1fea7] [ Links ]
13. Liang C, Needham M. Necrotizing autoimmune myopathy. Curr Opin Rheumatol 2011;23:612-619. [http://dx.doi.org/10.1097/BOR.0b013e32834b324b] [ Links ]
14. Hengstman GJD, ter Laak HJ, Vree Egberts WT, et al. Anti-signal recognition particle autoantibodies: Marker of a necrotising myopathy. Ann Rheum Dis 2006;65:1635-1638. [http://dx.doi.org/10.1136/ard.2006.052191] [ Links ]
15. Pestronk A. Acquired immune and inflammatory myopathies: Pathologic classification. Curr Opin Rheumatol 2011;23:595-604. [http://dx.doi.org/10.1097/BOR.0b013e32834bab42] [ Links ]
 Correspondence:
Correspondence:
M Manie
(mou@sun.ac.za

