










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSAMJ: South African Medical Journal
On-line version ISSN 2078-5135
Print version ISSN 0256-9574
SAMJ, S. Afr. med. j. vol.105 n.7 Pretoria Jul. 2015
http://dx.doi.org/10.7196/SAMJNEW.7917
RESEARCH
Identification of a mutation in the ubiquitin-fold modifier 1-specific peptidase 2 gene, UFSP2, in an extended South African family with Beukes hip dysplasia
C M WatsonI; L A CrinnionII; L GleghornIII; G A WallisIV; W G NewmanV; R RamesarVI; P BeightonVII
IBSc, PhD; Yorkshire Regional Genetics Service and School of Medicine, University of Leeds, St James's University Hospital, Leeds, UK
IIBSc; Yorkshire Regional Genetics Service and School of Medicine, University of Leeds, St James's University Hospital, Leeds, UK
IIIBSc, PhD; Wellcome Trust Centre for Cell-Matrix Research, University of Manchester, Manchester, UK
IVBSc, MA, PhD; Wellcome Trust Centre for Cell-Matrix Research, University of Manchester, Manchester, UK
VPhD, FRCP; Centre for Genomic Medicine, Manchester Academic Health Science Centre, University of Manchester and Central Manchester University Hospitals NHS Foundation Trust, Manchester, UK
VIBSc, MSc, MBA, PhD; MRC Human Genetics Research Unit, Division of Human Genetics, Institute for Infectious Diseases and Molecular Medicine, Faculty of Health Sciences, University of Cape Town, South Africa
VIIOMB, MD, PhD, FRCP, FRSSA; MRC Human Genetics Research Unit, Division of Human Genetics, Institute for Infectious Diseases and Molecular Medicine, Faculty of Health Sciences, University of Cape Town, South Africa
ABSTRACT
BACKGROUND: Beukes hip dysplasia (BHD) is an autosomal dominant disorder of variable penetrance that was originally identified in a large South African family of European origin. BHD is characterised by bilateral dysmorphism of the proximal femur, which results in severe degenerative osteoarthropathy. Previous studies mapped the disorder to a 3.34 Mb region on chromosome 4q35.
OBJECTIVE: To fine-map the BHD locus and identify the disease-causing mutation by direct sequencing.
RESULTS: The linked BHD allele was refined to 1.33 Mb, reducing the number of candidate genes from 25 to 16. Analysis of protein coding and invariant splice-site sequences in three distantly related individuals identified a single-candidate disease-causing variant c.868T>C within exon 8 of the ubiquitin-fold modifier 1 (Ufm1)-specific peptidase 2 gene, UFSP2. The presence of this unique mutation was confirmed in all 17 affected members of the BHD family who were genotyped. The mutation segregated with the BHD phenotype in the extended family with a two-point (single marker) LOD score of 10.4 (θ = 0.0 and 80% penetrance). The mutation predicts the substitution of a highly conserved amino acid, p.Tyr290His, in the encoded protein. In vitro functional assays performed using purified recombinant wild-type and mutant UFSP2 protein demonstrated that the BHD mutation abolishes UFSP2-mediated C-terminal cleavage of its substrate, Ufm1.
CONCLUSION: We report a unique UFSP2 mutation that segregates with the BHD phenotype. The predicted amino acid substitution inactivates UFSP2 proteolytic function, thus implicating the ubiquitin-fold modifier 1 cascade in this form of severe hip osteoarthropathy. The facile polymerase chain reaction-based assay we describe could be used to confirm the diagnosis of BHD, or for presymptomatic testing of members of the extended BHD family.
Skeletal development is controlled by extracellular matrix synthesis and deposition in combination with a complex regulatory network of cellular differentiation. Understanding these processes has been aided by mapping human disease genes, which have revealed fundamental insights into a number of the biological mechanisms underlying skeletogenesis. To date, more than 450 skeletal conditions have been identified and classified based on molecular, biochemical and/or radiographic criteria.[1] One such disorder is Beukes hip dysplasia (BHD) (OMIM #142669), a rare autosomal dominant condition that was originally identified in a large, multigeneration South African (SA) family of European descent.[2] BHD is characterised by severe progressive degenerative osteoarthritis (OA) of the hip joint in early adulthood. The condition is unique in that the underlying dysplasia and subsequent OA are confined to this region. Affected individuals are of normal stature and have no associated health problems. As described in detail previously, symptoms of hip joint discomfort usually develop in infancy or later childhood, but in a single individual initial presentation was as late as 35 years of age.[2] Phenotypic expression is age-related and variable in severity. The penetrance of this disorder is incomplete and has been estimated to be 80%. The earliest primary radiographic features of BHD include bilateral shortening and broadening of the femoral neck, delayed appearance of the secondary ossification centre, coxa vara, displacement of the femoral head in the acetabulum, and overgrowth of the greater trochanters. Following onset of symptoms, the characteristic signs of secondary OA (including bone sclerosis, cyst formation and narrowing of the joint space) develop and the joint deteriorates rapidly. We have previously mapped the BHD locus to a 3.34 Mb region of chromosome 4q35.[3] This locus has not been linked to other forms of familial acetabular dysplasia, indicating that BHD is a distinct disorder.[4,5]
To reduce the complexity of disease gene identification, we fine-mapped the chromosome 4q35 locus with polymorphic DNA markers. A combined Sanger and next-generation sequencing approach was used to screen coding and invariant splice-site sequences located within the linked interval for candidate pathogenic mutations. The development of 'next-generation sequencing' has accelerated the identification of disease-causing variants. This technology enables millions of DNA fragments covering all known protein coding and splice-site sequences of multiple individuals to be sequenced in a single experiment. This is typically achieved following a targeted enrichment reaction in which a sheared genomic DNA sample is hybridised to a pool of RNA baits that are complementary to the genomic region of interest. Through the application of this technology, we were able to confirm the presence of a single-candidate disease-causing variant within the BHD locus, which we subsequently characterised in vitro.
Methods
Archived DNA samples (extracted from blood samples obtained following informed consent for the purpose of molecular genetic analysis) were available from earlier investigations of the family, as reported previously.[3] A standard salting-out protocol was used to extract DNA from peripheral blood lymphocytes of 40 family members.
Fine-mapping the BHD locus
Microsatellite sizing and restriction fragment polymorphism genotyping of markers D4S1535 (Chr4: 185 235 750 - 185 236 098) and rs7663196 (Chr4: 186 570 521) was performed on members of the BHD family (Applied Biosystems, UK). Primer sequences are available on request. Two-point LOD scores were calculated using Linkage v5.1 (http://www.jurgott.org/linkage/LinkagePC.html).
Mutation screening and bioinformatic analysis
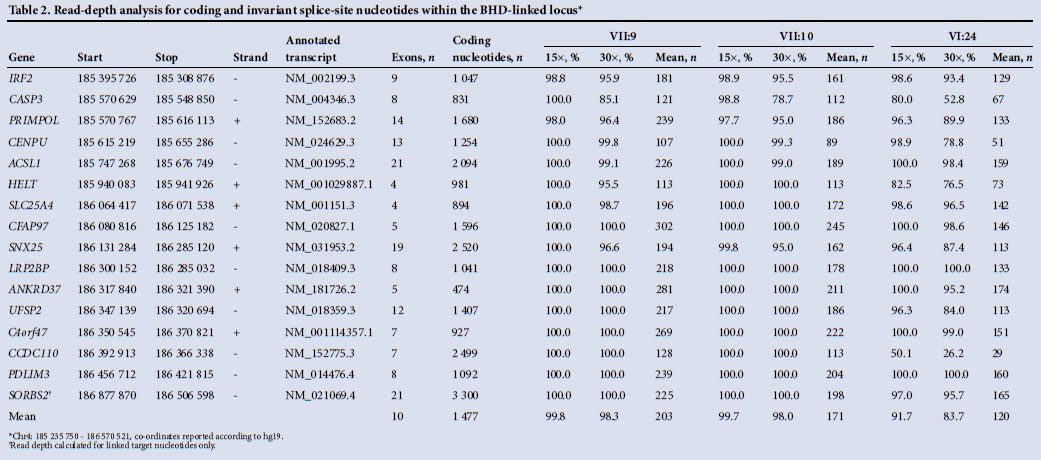
Comprehensive mutation screening of coding and flanking intron bases of three genes within the linked locus, CASP3, LRP2BP and UFSP2, was initially undertaken by Sanger sequencing (further details available on request). With the ubiquitous availability of next-generation sequencing, three distantly related individuals (VII:9, VII:10 and VI:24) were then selected for exome sequencing. Approximately 3 μg of genomic DNA was sheared using a Covaris S2 (Covaris Inc., USA) before standard whole-genome library preparation was performed, following the manufacturer's protocols throughout (Agilent Technologies, UK). Two enrichment polymerase chain reaction (PCR) reactions were necessary to obtain the required mass of genomic library for enrichment hybridisation, which was carried out using a SureSelect All Exon V5 (no UTRs) bait set. Final exome-enriched libraries were confirmed using an Agilent Bioanalyser prior to being pooled in equimolar concentrations. The pool was sequenced on a HiSeq2500 across three lanes of rapid mode flow cells in a run configuration that generated paired-end 100-bp reads (Illumina Inc., USA). Raw data were demultiplexed and converted to FASTQ.gz format using CASAVA v1.8.2. To analyse each individual, sequence reads were first aligned to an indexed human reference genome (hg19) using bwa v0.6.2. Duplicate reads were removed using Picard v1.85 (http://picard.sourceforge.net), and SAM/BAM file processing was performed using samtools v0.1.18 (http://samtools.sourceforge.net). GATKLite v2.3-4 was used to perform indel realignment, base quality recalibration, variant calling with the UnifiedGenotyper and read-depth analysis (http://www.broadinstitute.org/gatk/index.php). Variant call format files were annotated with position, frequency (incorporating dbSNP and the ESP5400 dataset) and in silico variant effect predictions using Alamut Batch v1.1.5 (http://www.interactive-biosoftware.com/software/alamut/overview). Interpretation of the annotated variant dataset was aided using AgileExomeFilter (http://dna.leeds.ac.uk/agile)[6] and the variant-decision support software Alamut Visual. To assess exon copy number variation, FishingCNV v2.1 (http://sourceforge.net/projects/fishingcnv/) was used to compare the sequenced read depth of BHD individuals to a pooled reference control comprising 65 BHD disease-free patients.
Molecular assay for UFSP2 mutation analysis
A 341-bp PCR amplicon spanning the UFSP2 c.868T>C mutation was generated using forward primer dCATTAAACATAATTCGGGAGCA and reverse primer dTCTGCACCATGAGGTAACAAA. Each PCR reaction consisted of 1 μL. of 15 ng/μL. DNA, 2.5 μl. of 10x PCR buffer, 1 μL of 50 mM MgCl2, 1 μl of 10 mM dNTPs, 0.5 μl of BIOTAQTM DNA polymerase (5 1%L), 17 μl of nuclease-free water, 1 μl of 10 μM forward primer and 1 μl of 10 μM reverse primer (Bioline Reagents Ltd., UK and Eurogentec !td., UK). Thermocycling conditions consisted of 95oC for 3 minutes followed by 36 cycles of 95oC for 30 seconds, 55oC for 45 seconds, 72oC for 45 seconds, and a final extension at 72oC for 10 minutes. The UFSP2 c.868T>C mutation introduced an NdeI (CATATG) restriction site that was detected by incubating PCR products with NdeI for 1 hour at 37oC and, if the mutation was present, generated 219-bp and 122-bp PCR fragments (NEB, UK). PCR products were resolved by tris-borate-EDTA agarose gel electrophoresis.
Functional analysis of the UFSP2 mutation
A previously described expression construct for mouse UFSP2 (NM_138668.2) was modified to incorporate the heterologous BHD mutation (c.844T>C, p.Tyr282His).[7] The wild-type (WT) (UFSP2 WT) and mutant (UFSP2 BHD) constructs together with a GST-Ufm1-HA construct were expressed in Escherichia coli and purified. Purified proteases were incubated with GST-Ufm1-HA at 37oC for 1 hour and the products were separated by SDS-PAGE gel electrophoresis (Invitrogen Ltd., UK). Protein bands were visualised by staining with Coomassie blue R-250.
Results
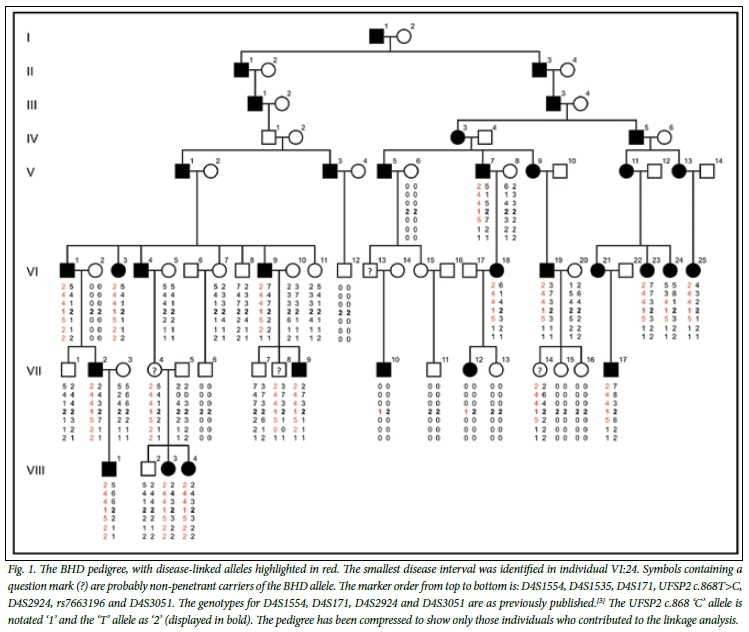
Since the original description of BHD in a multigeneration SA family, the pedigree has been extended to include family members in Canada, New Zealand and the UK. A condensed pedigree is shown in Fig. 1. A recent addition to the pedigree was affected individual VII:10, who was diagnosed at 31 years of age while resident in the UK. He presented with severe hip joint discomfort and pain that had increased progressively since the age of 13 years. Consistent with a diagnosis of BHD, his symptoms were confined to his hip joints; he was of normal stature, had no evidence of other deformity, and his general health was good. Specifically, he did not have joint hypermobility, cleft palate, or visual or hearing deficit. A skeletal survey revealed no evidence of abnormality other than in both of his hip joints, where the features were characteristic of BHD and associated OA was evident (Fig. 2). In keeping with the variable penetrance of the disorder, the progression of the clinical and radiological manifestations of VII:10 were less severe than those described for other family members. In addition, his father, although an obligate carrier of the BHD allele, was reported to be asymptomatic apart from mild joint discomfort at the age of 63 years.
BHD was previously linked to an 11-cM locus on chromosome 4q35.[3] Subsequently, we fine-mapped the locus using proximal and distal markers D4S1535 and rs7663196, respectively. Recombination events (see individual VI:24, Fig. 1) reduced the size of the linked region from 3.34 Mb to 1.33 Mb and decreased the number of candidate genes from 25 to 16. To identify the BHD mutation, systematic sequence analysis of the coding exons and intron splice sites of CASP3, LRP2BP and UFSP2 located in the linked region was conducted. This analysis identified a single novel heterozygous variant, c.868T>C, in UFSP2 exon 8 (NM_018359.3) (Fig. 3, A).



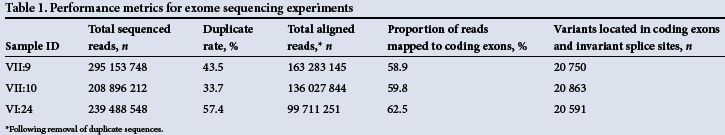
To ensure that no additional candidate disease-causing variants were present within the linked interval, whole-exome sequencing was performed on three distantly related individuals (VII:9, VII:10 and VI:24). Exome sequencing performance metrics (Table 1) demonstrated that the hybridisation capture efficiencies, as determined by the percentage of sequence reads mapping to each exome, were comparable to those reported elsewhere.[8] To determine the proportion of target nucleotides that were sufficiently well sequenced to exclude the presence of non-reference nucleotides, read-depth analysis was performed. For two of the three sequenced individuals, >98% of target bases had a read depth that was >30x, which is a conservative read-depth metric that has been widely adopted by the diagnostic sequencing community[9] (Table 2).
Variants with a reported minor allele frequency <0.2 located within the fine' mapped BHD locus (Chr4: 185 235 750 -186 570 521) were assessed for disease-related pathogenicity. This yielded a filtered dataset comprising one variant in individual VII:9, one in VII:10 and four in VI:24. The single variant found in common between the affected individuals was the UFSP2 c.868T>C variant that had been identified by the initial sequencing screen. This c.868T>C variant was not identified in publicly accessible variant databases including dbSNP and the ESP5400 dataset. In addition, exon-based copy number analysis did not identify any dosage variants within the linked interval in any of the three individuals.
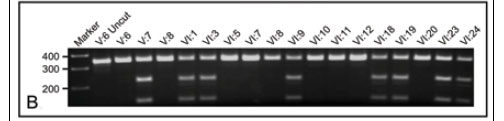
To explore familial segregation, all available members of the BHD family were genotyped for presence of the UFSP2 c.868T>C variant using PCR followed by restriction enzyme digestion with NdeI (see Fig. 3, B for a representative genotyping gel and Fig. 1 for the genotypes). All 17 individuals with a confirmed diagnosis of BHD were heterozygous for the c.868T>C mutation. There was evidence of non-penetrance in one obligate carrier (VII:4), and the mutation was found in two individuals (VII:8 and VII:14) in whom BHD had not been excluded or confirmed owing to their young age at time of consultation. Linkage analysis between BHD and the c.868T>C mutation generated a two-point (single marker) LOD score of 10.4 (at θ = 0.0 and 80% penetrance).
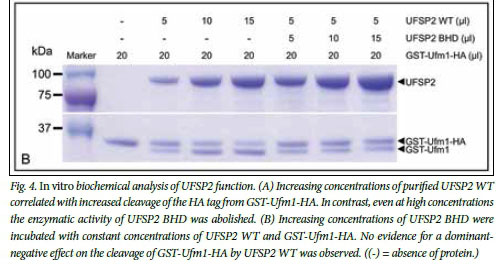
The UFSP2 c.868T>C variant predicts a tyrosine to histidine substitution at position 290 of the encoded protein, which is highly conserved across multiple species (Fig. 3, C). The effect of the p.Tyr290His substitution on UFSP2 protease activity was assayed in vitro as previously described by incubating recombinant purified mouse UFSP2 with Ufm1 that had been modified by the addition of an N-terminal GST tag and a C-terminal HA tag.[7] For this purpose, the heterologous mutation (c.844T>C, p.Tyr282His in the mouse) was inserted into the UFSP2 WT construct and expressed, and both the WT and UFSP2 BHD proteases were purified. Increasing concentrations of UFSP2 WT resulted in increased cleavage of the GST-Ufm1-HA tag (Fig. 4, A). In contrast, UFSP2 BHD did not cleave GST-Ufm1-HA, even at high concentrations. The processing activity of UFSP2 WT was not affected in the presence of increasing concentrations of UFSP2 BHD, indicating that UFSP2 BHD does not exert a dominant-negative effect at the level of Ufm1 processing in vitro (Fig. 4, B).

Discussion
BHD is a unique Mendelian disorder that has to date been described in only a single SA family of European origin.[2] The size and structure of the pedigree enabled the initial mapping of the linked locus to Chr4q35. [3] However, this locus did not contain any obvious candidate genes. To reduce the burden of variant interpretation following mutation screening, we fine-mapped the region and thereby reduced the number of candidate genes from 25 to 16. Initial mutation screening of genes located within the locus by Sanger sequencing identified a unique UFSP2 c.868T>C variant. This variant was identified in all individuals tested who had a confirmed diagnosis of BHD, but there was evidence of non-penetrance of BHD in the pedigree. The segregation of this variant with BHD generated a LOD score of 10.4 (at θ = 0.0 and 80% penetrance).
As no other forms of skeletal dysplasia had been mapped to the linked locus on Chr4q35, no other families with BHD had been reported and the UFSP2 c.868T>C variant appeared to be unique to the BHD family, independent verification that the variant was indeed the BHD mutation was not possible. We therefore undertook comprehensive exome analysis of the linked allele in three distantly related individuals. This analysis confirmed that there were no other candidate disease-causing mutations in linkage disequilibrium with UFSP2 c.868T>C, suggesting that this variant is the BHD mutation. The identification of this mutation will enable diagnostic accuracy of BHD, aid predictive testing of at-risk family members, and underpin future longitudinal studies aimed at correlating the progression of radiological features with the onset of clinical symptoms. The cysteine protease encoded by UFSP2 is highly conserved and is homologous to a second cysteine protease, UFSP1, that has a similar C-terminal domain but a shorter N-terminal domain.[7] Neither protease shares sequence homology to other identified proteases. UFSP1 and UFSP2 have been shown previously to cleave two C-terminal residues (S101 and C102) from the protein ubiquitin-fold modifier 1 (Ufm1).[7] Ufm1 is a post-translational modifier protein that is classified as a member of the family of ubiquitin-like proteins (Ubls). Following modification through an E1-E2-E3 multienzyme cascade, Ufm1 is attached to its target protein. The conjugation and deconjugation of target proteins by Ubls modulates their function and thereby the regulation of cellular processes.[10] In the Ufm1 modification pathway, Ufm1 is activated by either UFSP1 or UFSP2 to expose a C-terminal glycine residue.[7] Activated Ufm1 then reacts with Uba5 (E1-like enzyme) and is transferred to Ufc1 (E2-like enzyme) before being transferred to its target protein by Ufl1 (E3-like enzyme). At present, the full repertoire of Ufm1 target proteins remains to be identified. Although there are currently no reported studies detailing the tissue specificity or time course of UFSP2 expression, analysis of multiple mouse tissues has revealed that Ufm1 expression is abundant in protein-secreting cells.[11] Further, there is increasing evidence that the Ufm1 pathway has a role in the regulation of endoplasmic reticulum (ER) stress responses.[12] Interestingly, ER stress is a recognised pathogenic mechanism underlying a number of forms of osteochondrodysplasia, and the ER stress response has been proposed as a therapeutic target for such disorders.[13] This reported evidence therefore suggests a putative mechanistic link between the Ufm1/UFSP2 pathway and the BHD phenotype.
Alignment of multiple UFSP2 protein sequences from different multicellular organisms, including plants and animals, demonstrated that the p.Tyr290 residue has been conserved for at least 1.6 billion years.[14] In an in vitro assay we found that purified mouse UFSP2 containing the mouse equivalent of the p.Tyr290His BHD substitution did not cleave Ufm1, even at high concentrations. Lack of activity of the mutated protease is consistent with predictions made from the crystal structure of UFSP2 that the p.Tyr290 amino acid is a crucial residue within the UFSP2 active site.[15] In this in vitro assay, however, the mutated UFSP2 did not appear to exert a dominant-negative effect. Confirmation of the functional consequences of the BHD mutation on the Ufm1/UFSP2 pathway therefore requires further investigation.
Conclusion
In summary, genetic and functional data support that UFSP2 c.868T>C is the mutation causing BHD. The facile PCR-based assay that we have described could be used to confirm the diagnosis of BHD, or for presymptomatic testing of members of the extended BHD family.
References
1. Warman ML, Cormier-Daire V, Hall C, et al. Nosology and classification of genetic skeletal disorders: 2010 revision. Am J Med Genet A 2011;155A(5):943-968. [http://dx.doi.org/10.1002/ajmg.a.33909] [ Links ]
2. Cilliers, HJ, Beighton P. Beukes familial hip dysplasia: An autosomal dominant entity. Am J Med Genet 1990;36(4):386-390. 'http://dx.doi.org/10.1002/ajmg.1320360403] [ Links ]
3. Roby P, Eyre S, Worthington J, et al. Autosomal dominant (Beukes) premature degenerative osteoarthropathy of the hip joint maps to an 11-cM region on chromosome 4q35. Am J Hum Genet 1999;64(3):904-908. [http://dx.doi.org/10.1086/302291] [ Links ]
4. Ingvarsson T, Stefánsson SE, Gulcher JR, et al. A large Icelandic family with early osteoarthritis of the hip associated with a susceptibility locus on chromosome 16p. Arthritis Rheum 2001;44(11):2548- 2555. [http://dx.doi.org/10.1002/1529-0131(200111)44:11%3C2548::AID-ART435%3E3.0.CO;2-S] [ Links ]
5. Mabuchi A, Nakamura S, Takatori Y, Ikegawa S. Familial osteoarthritis of the hip joint associated with acetabular dysplasia maps to chromosome 13q. Am J Hum Genet 2006;79(1):163-168. [http://dx.doi.org/10.1086/505088] [ Links ]
6. Watson CM, Crinnion LA, Morgan JE, et al. Robust diagnostic genetic testing using solution capture enrichment and a novel variant-filtering interface. Hum Mutat 2014;35(4):434-441. [http://dx.doi.org/10.1002/humu.22490] [ Links ]
7. Kang SH, Kim GR, Seong M, et al. Two novel ubiquitin-fold modifier 1 (Ufm1)-specific proteases, UfSP1 and UfSP2. J Biol Chem 2007;282(8):5256-5262. [http://dx.doi.org/10.1074/jbc.M610590200] [ Links ]
8. Bodi K, Perera AG, Adams PS, et al. Comparison of commercially available target enrichment methods for next-generation sequencing. J Biomol Tech 2013;24(2):73-86. [http://dx.doi.org/10.7171/jbt.13-2402-002] [ Links ]
9. Weiss MM, van der Zwaag B, Jongbloed JD, et al. Best practice guidelines for the use of next-generation sequencing applications in genome diagnostics: A national collaborative study of Dutch genome diagnostic laboratories. Hum Mutat 2013;34(10):1313-1321. [http://dx.doi.org/10.1002/humu.22368] [ Links ]
10. Daniel J, Liebau E. The ufm1 cascade. Cells 2014;3(2):627-638. [http://dx.doi.org/10.3390/cells3020627] [ Links ]
11. Lemaire K, Moura RF, Granvik M, et al. Ubiquitin fold modifier 1 (UFM1) and its target UFBP1 protect pancreatic beta cells from ER stress-induced apoptosis. PLoS One 2011;6(4):e18517. [http://dx.doi.org/10.1371/journal.pone.0018517] [ Links ]
12. Zhang Y, Zhang M, Wu J, Lei G, Li H. Transcriptional regulation of the Ufm1 conjugation system in response to disturbance of the endoplasmic reticulum homeostasis and inhibition of vesicle trafficking. PLoS One 2012;7(11):e48587. [http://dx.doi.org/10.1371/journalpone.0048587] [ Links ]
13. Boot-Handford RP, Briggs MD. The unfolded protein response and its relevance to connective tissue diseases. Cell Tissue Res 2010;339(1):197-211. [http://dx.doi.org/10.1007/s00441-009-0877-8] [ Links ]
14. Wang DY, Kumar S, Hedges SB. Divergence time estimates for the early history of animal phyla and the origin ofplants, animals and fungi. Proc Biol Sci 1999;266(1415):163-171. [http://dx.doi.org/10.1098/rspb.1999.0617] [ Links ]
15. Ha BH, Jeon YJ, Shin SC, et al. Structure of ubiquitin-fold modifier 1-specific protease UfSP2. J Biol Chem 2011;386(12):10248-10257. [http://dx.doi.org/10.1074/jbc.M110.172171] [ Links ]
 Correspondence:
Correspondence:
C M Watson
c.m.watson@leeds.ac.uk
Accepted 11 June 2014.


{kind=link}
{kind=link}
{kind=link}