










Servicios Personalizados
Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkSAMJ: South African Medical Journal
versión On-line ISSN 2078-5135
versión impresa ISSN 0256-9574
SAMJ, S. Afr. med. j. vol.105 no.7 Pretoria jul. 2015
http://dx.doi.org/10.7196/SAMJNEW.7880
FORUM
GENETICS
A South African family with oculopharyngeal muscular dystrophy: Clinical and molecular genetic characteristics
C M SchutteI; C M DorflingII; R van CollerIII; E M HoneyIV; E J van RensburgV
IHead of the Department of Neurology in the Faculty of Health Sciences, University of Pretoria, South Africa. She has a special interest in genetic neurological conditions
IIResearch Officer in Lizette van Rensburgs Cancer Genetics Research Group
IIINeurologist in private practice who is affiliated to the Department of Neurology at the University of Pretoria
IVPaediatrician with a special interest in human genetics, has been working in the Department of Genetics for the past 15 years
VAssociate Professor of Human Genetics in the Department of Genetics, Faculty of Health Sciences, University of Pretoria, has been involved in molecular genetic research for the past 20 years, with a special interest in genetic susceptibility to cancer
ABSTRACT
Autosomal dominantly inherited oculopharyngeal muscular dystrophy (OPMD) is caused by a trinucleotide repeat expansion in exon 1 of the polyadenylate binding protein nuclear 1 (PABPN1) gene on chromosome 14q. A large family with OPMD was recently identified in Pretoria, South Africa (SA). Molecular studies revealed a (GCG)11(GCA)3GCG or (GCN)15 mutant allele. The (GCN)15 mutation detected in this family has been described previously in families from Uruguay and Mexico as a founder effect. To our knowledge, this is the first report of an SA Afrikaner family with molecularly confirmed OPMD. The proband, a 64-year-old woman, presented to the neurology outpatient department at Steve Biko Academic Hospital, Pretoria. A sibship of 18 individuals was identified, of whom eight had OPMD. Four patients were interviewed and examined clinically, and electromyographic studies were performed. Molecular analysis of the PABPN1 gene was performed by polymerase chain reaction amplification and direct sequencing of exon 1 in three of the patients. Patients presented with ptosis, external ophthalmoplegia, dysphagia, dysarthria and mild proximal weakness. High foot arches and absent ankle reflexes raised the possibility of peripheral neuropathy, but electromyography showed only mildly low sensory amplitudes, and myopathic units in two patients.
Oculopharyngeal muscular dystrophy (OPMD) (OMIM #164300) is a late-onset (>45 years) myopathy characterised by progressive ptosis, dysphagia and varying degrees of proximal muscle weakness. The ptosis is mostly restricted to the levator palpebrae and later involves other extraocular muscles. Complete external ophthalmoplegia is rare. The dysphagia is initially for solids only, but steadily progresses to an extent that patients may become malnourished. In later life these patients may suffer from bouts of aspiration pneumonia. Although proximal muscle weakness, facial weakness and dysarthria may occur, smooth and cardiac muscles appear to be spared. Pathologically, intranuclear inclusions are observed in the muscle fibres.[1]
Early descriptions of families with ptosis and dysphagia drew attention to the hereditary basis of the disorder, and after the 1962 publication by Victor et al.[2]the condition was recognised as a form of muscular dystrophy and given the name OPMD. The condition is mainly inherited in an autosomal dominant manner, although uncommon recessive forms have been described.[1]
OPMD has been recognised as a trinucleotide repeat expansion disorder.[3] The PABN1 gene, also known as PABP2, encoding the polyadenylate binding protein nuclear 1, has a polyalanine tract in the N-terminal end. The wild-type allele is (GCG)6(GCA)3GCG, adding up to a total of ten alanine residues (GCNs). Abnormal expansion of this tract results in OPMD.[3] The expansion usually occurs in the (GCG)6 repeat, but repeats of the (GCA)3 have been documented. Individuals with OPMD have expansions ranging from 12 to 17 GCNs.[1] Normal PABN1 is a nuclear protein that is involved in the polyadenylation of messenger RNA.[4] It associates with RNA polymerase II during transcription and facilitates movement of the released transcript through the nuclear pore, thus acting as a molecular chaperone for the proper export of poly(A) RNA from the nucleus.[5] Additionally, PABN1 was found to bind to SKIP (ski-interacting protein), and together they directly control the expression of muscle-specific genes. Following on from this, Apponi et al.[6]demonstrated that the normal protein plays an important role in myoblast proliferation and differentiation, whereas the extended polyalanine tract causes clumping or the PABN1 protein and accumulation in the nuclei of the skeletal muscle fibres.[7] Recently Davies and Rubinsztein[8] reported that apoptosis is directly involved in the pathology of OPMD, and that it is a major contributor to the muscle dysfunction in the disease.
The incidence of OPMD varies widely, ranging from 1/200 000 in France to 1/600 in Israel (Bukhara Jewish population). The disease has been described in more than 35 countries worldwide, with some variations in clinical presentation.
To our knowledge, this is the first South African (SA) OPMD family to be described. We report on the clinical and molecular genetic characteristics of this family.
The proband was a 64-year-old woman who presented with ptosis, progressive dysphagia and some proximal weakness to the neurology outpatient department at Steve Biko Academic Hospital in Pretoria, SA. With the assistance of the patient, a family pedigree was drawn up and contact details of as many family members as possible were obtained. All were then contacted and invited to take part in this study. After informed consent had been obtained, four individuals were subsequently interviewed and examined clinically, and had electromyographic (EMG) studies performed. In addition, blood samples were taken from three individuals.
Genomic DNA was extracted from peripheral blood using standard protocols. Exon 1 of PABN1, containing the region where the GCN repeat is located, was amplified using primers previously described[9] with the Failsafe PCR Enzyme mix with buffer J (Epicentre Biotechnologies, USA). Cycle sequencing of the amplicons was performed with BigDyeV3.1 (Applied Biosystems, USA) and analysed on an ABI 3130 (Applied Biosystems).
The study was approved by the Ethics Committee of the Faculty of Health Sciences, University of Pretoria.
Family pedigree
An abbreviated pedigree of this Afrikaner family is depicted in Fig. 1. The index patient (III:17) was one of a sibship of 18, eight of whom are affected with OPMD. Their mother (II:2) was affected from the age of approximately 40 years. Noteworthy is the offspring of III:2, an unaffected daughter, who happened to marry her maternal cousin (III:3) who had OPMD. Their children (IV:1 and IV:2) inherited the disorder from their father.
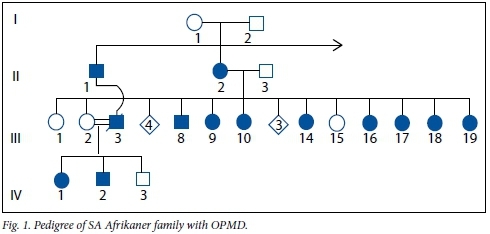
Patient III:17
This 64-year-old woman reported that her first symptoms of ptosis had started at the age of about 40 years, but really interfered with her vision only 10 years later, when she needed eyelid surgery to correct the ptosis. Dysphagia was also present from age 40 years and progressed slowly up to a point where she now has to be very selective about the food she eats. On examination, she had normal higher functions (Mini Mental State Examination (MMSE) 27/30). She had dysarthria with somewhat nasal speech and often had to clear her throat while speaking. The range of eye movements was restricted, especially upwards in that she could not elevate her eyes beyond the neutral position. Ptosis was not prominent after the eyelid surgery. Mild facial weakness was present, as well as weakness of neck flexion (4/5) and mild proximal weakness in the upper and lower limbs. Deep tendon reflexes could only be elicited with augmentation, but ankle reflexes were absent. The patient had high-arched feet, but the findings on sensory examination were unremarkable.
On EMG, peroneal, tibial and radial motor responses were normal. The sural response showed slightly low amplitude, as did the medial and lateral plantar responses. The peripheral autonomic sensory potentials (PASPs) were present, and the response to repetitive stimulation of the ulnar nerve was within normal limits. A needle examination showed normal motor units in the deltoid muscle.
Patient III:9
This woman was 75 years old when examined. Her data were retrieved retrospectively from her neurologist's patient record files because she lived far away. She had severe dysphagia and had lost 15 kg over the past few months. On examination, she had an almost total external ophthalmoplegia and ptosis, more marked on the left. There was weakness of the soft palate and tongue, and her speech was hoarse and dysarthric. Mild proximal and distal weakness was present, but this was difficult to interpret owing to her weak general condition. Findings on EMG conduction studies of the median and ulnar motor and sensory nerves were normal. Myopathic units were found in the deltoid muscle, and needle examination of the tongue revealed normal insertional activity and normal motor unit potentials.
The findings on repetitive stimulation studies of the accessory nerve were also normal. The patient underwent insertion of a gastrostomy tube to facilitate feeding.
Patient III:14
As with III:17, this 70-year-old woman's ptosis had started at approximately 40 years of age. She had experienced the first symptoms of dysphagia at around 50 years, after which it progressively worsened. Eyelid repair surgery had been performed twice in the past 20 years. On examination, higher mental functions were normal (MMSE 28/30), but the patient had marked dysarthria with prominent nasal speech and lip weakness; the eye movements were full except for some mild restriction of lateral gaze to both sides. There was asymmetry on elevation of the soft palate and severe dysphagia. She had a clearly myopathic face with weakness of the frontalis, levator palpebrae superioris and orbicularis oculi muscles and severe weakness of the upper lip levators and levator anguli oris. Limb weakness was very mild: the deltoids, pectorals, biceps, triceps and hip flexors and extensors were grade 4+/5 and, interestingly, foot dorsiflexion and finger extension was also weak at grade 4/5. The deep tendon reflexes of the upper limbs were only present as a flicker with augmentation manoeuvres, the patella reflex was 2+/4, and the ankle reflexes were absent. The findings on sensory examination were normal. The patient also had high-arched feet.
EMG showed normal responses of the peroneal, tibial and radial motor nerves, the medial and lateral plantar responses showed low amplitudes, and the sural responses were normal. The PASPs were present and the response to repetitive stimulation of the ulnar nerve was normal. A needle examination of the left deltoid muscle showed some small myopathic units but no fibrillation potentials.
Patient III:18
Consistent with her sisters, this 63-year-old woman had noted ptosis at the age of 40 years and started complaining of dysphagia approximately 10 years later. She had had eyelid repair operations at least 15 years previously, and had also had oesophageal sphincter botulinum toxin injections and myotomy for severe dysphagia at the age of 56. She did not complain of limb muscle weakness. However, she noted that she had felt tired after bypass surgery following a myocardial infarction 2 months prior to her visit to neurology. On examination, her higher mental functions were normal (MMSE 30/30). She had very mild limitation of upward and lateral gaze, and a mild dysarthria was noticeable. Orbicularis oculi muscle power was slightly decreased and mild ptosis was present. Power of the latissimus dorsi and hip extensors was mildly reduced (4+/5), deep tendon reflexes in the upper limbs were a flicker with augmentation, the patella reflex was 2+/4, and the ankle reflexes were absent bilaterally. Findings on sensory examination and the co-ordination and gait were normal.
On EMG, the peroneal, tibial and radial motor responses as well as the response to repetitive stimulation of the ulnar nerve were normal, and PASPs were present. The amplitude of the medial and lateral plantar responses was somewhat decreased, but conduction velocities were normal. The amplitude of the sural response was also slightly decreased at point A. A needle examination showed normal muscle unit potentials in the deltoid muscle.
Molecular analysis
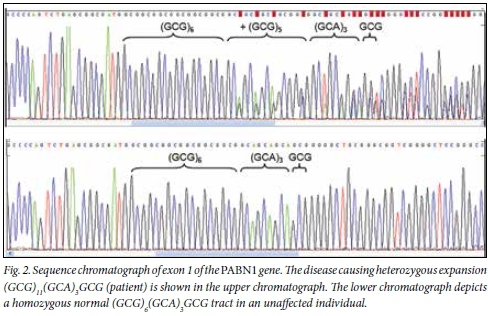
As shown in Fig. 2, cycle sequencing of exon 1 of the PABPN1 gene of the three patients (III:14, III:17 and III:18) from whom blood samples were available revealed that they were heterozygous for a (GCG)11(GCA)3GCG or (GCN)15 mutant allele. This expansion therefore increases the total number of alanine residues (GCNs) from the normal ten to 15.
Discussion
We describe the first SA Afrikaner family with genetically proven OPMD. The Afrikaners are mainly descended from Dutch, German and some French immigrants to the Cape during the 17th century. It is estimated that the founding Afrikaner population consisted of approximately 90 families by 1687. We therefore expected to find that our family would share a mutation with one of these founding populations. Many individuals with OPMD from North America and Europe carry a mutant (GCG)9 or (GCN)13 allele.[3] Studies from the UK showed an equal distribution of (GCG)9 and (GCG)10 (or (GCN)13 and (GCN)14) mutations,[10] and in Hispanic New Mexicans, the (GCG)9 mutation was also commonly identified.[11]
Our SA Afrikaner family has the (GCG)11(GCA)3GCG (or (GCN)15) mutation, which has been established to be a founder mutation in Uruguayan and Mexican families.[12] Both the genealogical and molecular data suggested that the ancestors of the Uruguayan population with OPMD were settlers in the Canary Islands in the 19th century,[12] with the possibility that the mutation arose between the 10th and 14th centuries in the Old World. In the Mexican population, the possibility of two independent founder effects has been suggested, since the (GCG)11(GCA)3GCG (or (GCN)15) and (GCG)9 or (GCN)13 mutant alleles both occurred in the investigated patients.[13] Interestingly, in the study from Uruguay, one of the patients was an individual from SA who also carried the (GCG)11(GCA)3GCG (or (GCN)15) mutation, but no further details on this patient are available.
The patients in the current study presented with typical symptoms and signs of autosomal dominant OPMD. Ptosis started around the age of 40 years, closely followed by progressive dysphagia. Corrective surgery for ptosis was performed on three patients, and one also had oesophageal surgery. The mutant (GCG)11(GCA)3GCG (or (GCN)15) allele may be associated with an earlier onset of symptoms, specifically ptosis, than the mutant (GCG)9 or (GCN)13 allele. Our patients reported an onset of ptosis at around 40 years, which has also been described in the Mexican population, where the mean age at onset of ptosis was 46.5 years in subjects with the (GCG)11(GCA)3GCG (or (GCN)15) mutation, compared with 54.7 years in those with the (GCG)9 or (GCN)13 mutation.[13] All patients showed signs of external ophthalmoplegia, which was severe in two cases.
Additionally, all patients had dysarthria, which ranged from very mild to moderate. Dysarthria is a finding that has been reported in patients with OPMD, but is rarely emphasised. A study by Young and Durant-Jones[14] evaluated voice intensity, resonance and pitch range in five patients with OPMD, showing changes in voice, articulation and resonance in all individuals, which was worse in those older than 70 years. Hyper-nasality, as a finding of the dysarthria, was reported in three of their patients. Of our four patients, the older two, at 70 and 75 years, also had the worst dysarthria. Mild proximal weakness, often described in OPMD, was noted in all our patients and neck flexion weakness in two. Interestingly, two of our patients also showed mild weakness of the tibialis anterior muscles, and one of the older patients also had clear weakness of finger extension, uncommonly seen in OPMD. The high foot arches in two patients and the absent ankle reflexes raised the clinical possibility of a peripheral neuropathy.
On electrophysiological studies, clear myopathic units were seen in two of our patients, and findings on all motor nerve conduction studies were normal. Low amplitudes of the medial and lateral plantar responses were noted in the three patients in whom these were tested, but the abnormalities were mild; in two, the amplitudes of the sural nerve responses were also minimally reduced. Involvement of the peripheral nerves in OPMD has been debated in the past decade. Finsterer[15] concluded in a recent editorial that involvement of peripheral nerves in OPMD should be evaluated further by studying only patients with genetically confirmed OPMD, since many of the patients with electrophysiological abnormalities were reported in the pre-genetic era. The significance of our electrophysiological findings is unclear; motor nerves and autonomic responses were not involved electrophysiologically, and if anything the lower amplitudes of the plantar responses may indicate a mild sensory neuropathy. However, all our patients were older than 60 years, age perhaps influencing the amplitude of sensory nerve responses, although the medial plantar responses should not be absent before the age of 70 years.
Conclusion
In conclusion, we have described the clinical and genetic features of the first SA family with OPMD.
Author contributions. CMS initiated the project, interviewed the patients, examined them and wrote up the clinical part of the paper; CMD and EJvR did the genetic analysis and wrote the genetic part of the article; and EMH and RvC helped with the clinical management of patients and reviewing of the manuscript.
References
1. Brais B. Oculopharyngeal muscular dystrophy: A polyalanine myopathy. Curr Neurol Neurosci Rep 2009;9(1):76-82. [http://dx.doi.org/10.1007/s11910-009-0012-y] [ Links ]
2. Victor M, Hayes R, Adams RD. Oculopharyngeal muscular dystrophy: A familial disease of late life characterized by dysphagia and progressive ptosis of the eyelids. N Engl J Med 1962;267(25):1267-1272. [http://dx.doi.org/10.1056/NEJM196212202672501] [ Links ]
3. Brais B, Bouchard J-P, Xie Y-G, et al. Short GCG expansions in the PABP2 gene cause oculopharyngeal muscular dystrophy. Nat Genet 1998;18(2):164-167. [http://dx.doi.org/10.1038/1304] [ Links ]
4. Wahle E, Lustig A, Jeno P, Maurer P. Mammalian poly(A)-binding protein II: Physical properties and binding to polynucleotides. J Biol Chem 1993;268(4):2937-2945. [ Links ]
5. Bear DG, Fomproix N, Soop T, Bjorkroth B, Masich S, Daneholt B. Nuclear poly(A)-binding protein PABN1 is associated with RNA polymerase II during transcription and accompanies the released transcript to the nuclear pore. Exp Cell Res 2003;286(2):332-344. [http://dx.doi.org/10.1016/S0014-4827(03)00123-X] [ Links ]
6. Apponi LH, Leung SW, Williams KR, Valentini SR, Corbett AH, Pavlath GK. Loss of nuclear poly(A)-binding protein 1 causes defects in myogenesis and mRNA biogenesis. Hum Mol Genet 2010;19(6):1058-1065. [http://dx.doi.org/10.1093/hmg/ddp569] [ Links ]
7. Uyama E, Tsukahara T, Goto K, et al. Nuclear accumulation of expanded PABP2 gene product in oculopharyngeal muscular dystrophy. Muscle Nerve 2000;23(10):1549-1554. [http://dx.doi.org/10.1002/1097-4598(200010)23:10<1549::AID-MUS11>3.0.CO;2-0] [ Links ]
8. Davies JE, Rubinsztein DC. Over-expression of BCL2 rescues muscle weakness in a mouse model of oculopharyngeal muscular dystrophy. Hum Mol Genet 2011;20(6):1154-1163. [http://dx.doi.org/10.1093/hmg/ddq559] [ Links ]
9. Bae JS, Ki C-S, Kim J-W, Kim BJ. Identification of a novel mutation in a Korean patient with oculopharyngeal muscular dystrophy. J Clin Neurosci 2007;14(1):89-92. [http://dx.doi.org/10.1016/j.jocn.2005.12.036] [ Links ]
10. Hill ME, Creed GA, McMullan TF, et al Oculopharyngeal muscular dystrophy: Phenotypic and genotypic studies in a UK population. Brain 2001;124(3):522-526. [http://dx.doi.org/10.1093/brain/1243.522] [ Links ]
11. Becher MW, Morrison L, Davis LD, et al. Oculopharyngeal muscular dystrophy in Hispanic New Mexicans. JAMA 2001;286(19):2437-2440. [http://dx.doi.org/10.1001/jama.286.19.2437] [ Links ]
12. Rodriguez M, Camejo C, Bertoni B, et al. (GCG)11 founder mutation in the PABPN1 gene of OPMD Uruguayan families. Neuromuscular Disorders 2005;15(2):185-190. [http://dx.doi.org/10.1016/j.nmd.2004.10.012] [ Links ]
13. Rivera D, Mejia-Lopez H, Pompa-Mera EN, et al Two different PABPN1 expanded alleles in a Mexican population with oculopharyngeal muscular dystrophy arising from independent founder effects. Br J Ophthalmol 2008;92(7):998-1002. [http://dx.doi.org/10.1136/bjo.2007.131482] [ Links ]
14. Young EC, Durant-Jones L. Gradual onset of dysphagia: A study of patients with oculopharyngeal muscular dystrophy. Dysphagia 1997;12(4):196-201. [http://dx.doi.org/10.1007/PL00009536] [ Links ]
15. Finsterer J. Involvement of the peripheral nerves in oculopharyngeal muscular dystrophy. Clin Neurophysiol 2010;121(6):803-804. [http://dx.doi.org/10.1016/j.clinph.2010.01.022] [ Links ]
 Correspondence:
Correspondence:
C M Schutte
Clinical data
cschutte@medic.up.ac.za
E J van Rensburg
Genetic data
lizette.jansenvanrensburg@up.ac.za
Accepted 6 November 2014.

