










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSAMJ: South African Medical Journal
On-line version ISSN 2078-5135
Print version ISSN 0256-9574
SAMJ, S. Afr. med. j. vol.105 n.4 Pretoria Apr. 2015
http://dx.doi.org/10.7196/SAMJ.9532
CONTINUING MEDICAL EDUCATION
REVIEW
Paediatric chronic kidney disease
I van BiljonI; A M MeyersII
IMMed (Paed), FC Paed (SA), Cert Nephrology (SA) Paed; Department of Paediatrics, Faculty of Health Sciences, University of Pretoria, South Africa
IIMB BCh, FCP (SA), Cert Nephrology (SA), FRCP (Lond); Donald Gordon Medical Centre, Klerksdorp Hospital, and National Kidney Foundation of South Africa, Johannesburg, South Africa
ABSTRACT
Doctors use various guidelines on paediatric chronic kidney disease (CKD) for managing their patients according to the availability of resources. As with adolescent and adult patients, CKD in children can also progress to end-stage renal failure - the time course being influenced by several modifiable factors. Decline in renal failure is best categorised in stages, which determine management and prognosis. Staging is based on three categories, i.e. cause, glomerular filtration rate and proteinuria. Early diagnosis of CKD allows for the institution of renoprotective treatment of modifiable factors and treatment to prevent the development of complications. The two most important modifiable factors that can be treated successfully are hypertension and proteinuria.
The objective of this article is to provide information on the diagnosis and treatment of CKD in children. Early identification and treatment of modifiable risk factors of CKD decreases the burden of disease and delays or prevents the need for renal replacement therapy.
Nephrology is a subspecialty often associated with expensive and technologically advanced therapies. Furthermore, paediatric nephrology services are limited as there are only a few registered paediatric nephrologists in South Africa (SA), almost all of whom practise in tertiary academic hospitals distributed unevenly across the country. The objective of this article is to provide information to general practitioners and paediatricians caring for children with chronic kidney disease (CKD).
The Kidney Disease Improving Global Outcomes guideline proposes the following definition of CKD:
- Abnormalities of kidney structure or function that have been present >3 months, with implications for the health of the patient, and either of the following:
- decreased glomerular filtration rate (GFR) <60 mL/min/1.73 m2, and/or
- presence of markers of kidney damage.
- Markers of kidney disease include any of the following:
- proteinuria (urine dipstick >2+ or urine protein:creatinine ratio 5 x the upper limit of normal)
- urine sediment abnormalities
- electrolyte and other abnormalities caused by tubular disorders
- histological abnormalities
- structural abnormalities detected by imaging
- history of renal transplantation.
The aforementioned definition does not apply to children <2 years of age in whom an appropriate age-adjusted GFR value should be used instead of GFR <60 mL/min/1.73 m2. Mature adult renal function is only achieved at 2 years of age. In infants <3 months old, a duration of >3 months does not apply.
Causes of CKD in children
Causes of CKD can be primary, i.e. no systemic disease present, or secondary, i.e. systemic disease affecting the kidney.
The most common causes of CKD in children are:
- congenital abnormalities of the kidney and urogenital tract (CAKUT), e.g. obstructive uropathy and reflux nephropathy
- hypoplastic/dysplastic kidneys
- primary focal and segmental glomerulo-sclerosis (nephrotic syndrome)
- haemolytic uraemic syndrome
- immune complex glomerulonephritis/ chronic glomerulonephritis
- hereditary nephropathies, e.g. polycystic kidney disease
- other rarer causes, e.g. drug/toxin-related renal disease.
Glomerular filtration rate category
In children the estimated GFR (eGFR) is calculated using the modified Schwartz formula:
[40 x height (cm)] 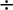 serum creatinine (Hmol/L) (ml/min/1.73 m2).
serum creatinine (Hmol/L) (ml/min/1.73 m2).
Table 1 lists the GFR categories.

Proteinuria category
A positive urine dipstick test for proteinuria should be confirmed with quantitative proteinuria measurement (measured as urine:protein:creatinine ratio (g/mmol) on a spot urine sample). Table 2 shows the proteinuria categories applicable to children.

Proteinuria is influenced by age, e.g. infants usually have a higher urinary loss of both glomerular and tubular protein, underlying pathology, and stage of CKD. Proteinuria eventually develops in all patients with CKD and is both a marker and a risk factor for the progression of this condition.
Symptoms and signs of CKD
Acquired CKD is generally a silent disease, which may only become symptomatic in its more advanced stages. Symptoms are often nonspecific, e.g. infants usually present with feeding-related complaints. Poor growth is the single most common, and almost universal, feature in all children.
In infants with CAKUT, the disease may be discovered by routine antenatal ultrasound scanning during pregnancy. In older children, the diagnosis may come as a complete surprise when a pathological condition is discovered co-incidentally.
Medical history
This should start with the antenatal history and include the perinatal and infant periods and other preceding illnesses and investigations, and concentrate on current symptoms of all the major organ systems.
Of importance is the history of the antenatal period with regard to CAKUT in the fetus, including oligohydramnios, maternal disease (e.g. hypertension (HT), infections, diabetes mellitus), and drug exposure (e.g. nonsteroidal anti-inflammatory drugs (NSAIDs) and angiotensin-converting enzyme (ACE) inhibitors).
Routine antenatal ultrasound scanning in the maternal and fetal unit is the most important diagnostic tool to diagnose CAKUT before the infant becomes symptomatic. Ideally, every woman should have at least one antenatal ultrasound scan during pregnancy, which is the practice in developed countries.
Kidney disease presents with nonspecific complaints, which contributes to a delay in making the diagnosis. Parents are often unable to recall the onset of symptoms because of the insidious nature of the disease.
A family history of kidney disease or specific pathology in other systems associated with inherited kidney disease, such as deafness, ear and eye abnormalities and HT, may give an indication of the diagnosis.
Examination
Clinical features associated with CKD
Longitudinal assessment of weight, length/ height and skull circumference plotted on an appropriate growth chart, is most important when evaluating a child's health. A complete physical examination should be done, including measurement of blood pressure (BP). HT in children is defined as systolic and/or diastolic BP >95th percentile for age, gender and height percentile. Ideally, one should use a BP percentile chart to interpret the BP level. Alternatively, a formula may be used, which provides a value corresponding to the 95th percentile BP for children of both sexes falling on the 50th percentile for height:
- systolic BP (95th percentile) - 1 - 17 years old: 100+ (age in years x 2)
- diastolic BP (95th percentile) - 1 - 10 years old: 60+ (age in years x 2)
- diastolic BP (95th percentile) - 11 - 17 years old: 70+ (age in years).
Other presenting features include:
- dysmorphic features typical of a syndrome associated with CAKUT, e.g. Eagle-Barrett syndrome
- tachypnoea mimicking acute 'respiratory distress' to compensate for metabolic acidosis
- chronic anaemia
- bone mineral disease, i.e. bone pain and skeletal deformities
- volume overload: oedema, HT, heart failure, pulmonary oedema
- uraemic symptoms and signs: nausea, vomiting, pruritus, brownish skin pigmentation, uraemic frost
- bleeding tendency (mucosa)
- convulsions due to HT, hyponatraemia, hypernatraemia, hypocalcaemia or uraemia.
Special investigations
- A urine dipstick test is an invaluable tool for the diagnosis of glomerular disease.
- A urine dipstick test does not detect tubular proteinuria, in which case urine amino acids or the β2-microglobulin:creatinine ratio should be measured.
- If proteinuria is present, the protein:creatinine ratio should be measured in an early-morning urine sample (normal <0.02 g/mmol).
- If a urine dipstick test reveals leucocytes and nitrites, a sterile sample of urine should be obtained for microscopy, culture and sensitivity.
- Twenty-four-hour urine or timed urine collection for creatinine clearance is impractical in children.
- Microscopy done on urine sediment provides information on the presence of dysmorphic red blood cells and red cell casts, which are diagnostic for glomerulonephritis.
- Serum urea, creatinine and electrolytes. Serum creatinine is necessary to calculate GFR. Serum urea is a poor tool to evaluate renal function, as it is influenced by hydration, nutritional status and protein intake.
- Urine volume may be normal or increased (polyuria) (>4 mL/kg/hour), or decreased (oliguria) (<1.0 mL/kg/hour).
- Serum calcium, phosphate, alkaline phosphatase and plasma parathyroid hormone should be determined to assess bone mineral disease and secondary hyperparathyroidism.
- Renal ultrasound is necessary to demonstrate kidney size (small shrunken kidneys are characteristic of CKD) and exclude urinary tract obstruction.
- There is no place for renal biopsy in patients with end-stage renal failure (ESRF).
Treatment
The principal renoprotective strategies with regard to modifiable risk factors for progression of CKD are the following:
- Treat and control HT and fluid overload. HT and cardiovascular disease are the two most common causes of death in children with CKD.
- Treat proteinuria with an ACE inhibitor, even in the absence of HT. Minimise proteinuria, using maximum tolerable doses of ACE inhibitors.
- Control hyperphosphataemia and hyper-parathyroidism and avoid hypercalcaemia.
- Treat anaemia.
- Treat dyslipidaemia.
- Investigate and treat infections.
- Provide a balanced nutritious diet to allow for optimal growth.
Monitoring and follow-up of children with CKD
Intervals between follow-up visits depend on the age of the child, staging of CKD and presence of complications. Children with stage 1 and 2 CKD are best followed up once every 6 months. Those with stage 3 and 4 CKD need to be followed up every 2 - 3 months.
Physical assessment at each visit should include:
- A complete physical examination. Monitor for end-organ damage of CKD.
- Monitor anthropometry and assess nutritional status.
- Monitor BP and cardiovascular system for volume overload, left ventricular hypertrophy/dilatation.
- Monitor mineral and bone disorder, looking for muscle weakness, delayed closure of the anterior fontanelle, bowing or deformities of the long bones.
- Assess neurodevelopment, especially in infants and children <5 years old.
- Fundoscopic examination must be done in all children with HT.
HT
The degree of HT correlates with severity of the underlying renal disease.
- Use long-acting drugs, preferably once daily.
- Evening administration may be more beneficial.
- Control BP to <90th BP percentile.
- In children with proteinuria, lower-target BP is recommended: 50 - 75th BP percentile as long as the child does not have symptoms of hypotension.
ACE inhibitors
- When proteinuria is present, an ACE inhibitor is the drug of choice for the treatment of HT.
- Beneficial effects include: decreasing BP, decreasing proteinuria independent of its effect on systemic BP, and slowing down the progression of CKD.
- ACE inhibitors should be administered over a run-in period.
- Enalapril 0.1 mg/kg/dose should be administered once daily.
- The dose may be increased to 0.5 mg/kg/day as a single dose or two divided doses.
- ACE inhibitor treatment is also indicated in all children with persistent nephrotic range proteinuria and a GFR >30 mL/min/1.73 m2, even in absence of HT.
Monitoring treatment
- ACE inhibitors may cause hyperkalaemia, worsen metabolic acidosis or cause decline in renal function.
- It is obligatory to confirm tolerability and monitor serum potassium, bicarbonate, and renal function within 5 - 7 days after the start of treatment, and regularly thereafter as indicated by the level of renal function.
- If the serum creatinine increases by >25%, hydration status should be checked, diuretics discontinued and dose of ACE inhibitor halved.
- If renal function does not improve, or hyperkalaemia of >5.5 mmol/L persists, discontinue ACE inhibitors.
Calcium channel blockers (CCBs)
- CCBs do not have renoprotective effects other than controlling BP.
- They can increase proteinuria, and are best used in non-proteinuric patients or in combination with ACE inhibitor/angiotensin II receptor blocker treatment.
- Amlodipine 0.1 - 0.3 mg/kg/day as a single daily dose (maximum dose 10 mg/day).
β-blockers
- β-blockers decrease pulse rate, cardiac output, afterload and renin release.
- Administer atenolol 1 - 2 mg/kg as a single daily dose or in two divided doses per day.
Other antihypertensive drugs
If the BP remains uncontrolled, review treatment compliance. In most cases uncontrolled HT is caused by volume overload, a direct consequence of non-compliance relating to a low salt diet and drug treatment.
CKD - bone and mineral disorder
CKD is associated with progressive phosphate retention, secondary hyperparathyroidism and development of bone mineral disease.
Management
Management includes dietary restriction of phosphate, phosphate binders and activated vitamin D.
Phosphate binders
- These should be used in combination with dietary phosphate restriction.
- Most of dietary phosphate originates from protein-containing foods.
- Restrict intake of dairy, grains and cereals, soft drinks, and food containing baking powder.
- Start phosphate binders early when the eGFR <70 mL/min/1.73 m2 or serum phosphate >1.8 mmol/L.
- Maintain parathyroid levels within the normal range.
Types of phosphate binders
These include:
- calcium carbonate, calcium acetate and sevelamer
- calcium carbonate (Titralac) 1 - 4 tablets three times daily with meals
- calcium acetate and sevelamer (Renagel) are more expensive and not commonly used in children.
Hydroxylated vitamin D (Ια-hydroxycholecalciferol or 1,25 dihydroxycholecalciferol) therapy
CKD is associated with a decreased production of activated vitamin D.
- Activated vitamin D should be given to all children with CKD with hypocalcaemia (serum calcium <2.2 mmol/L).
- If the serum phosphate is >2.5 mmol/L, hyperphosphataemia should be treated first to decrease serum phosphate to <1.8 mmol/L to prevent calcification of vascular and soft tissues.
- Start with α-calcidiol 0.25 μg/dose initially twice weekly.
- Increase the dose as necessary to maintain serum calcium in the upper normal range.
- Doses as high as 0.5 μg twice daily may be required.
Anaemia
Anaemia is defined as a haemoglobin (Hb) level of <11 g/dL in children 6 months - 5 years, <11.5 g/dL in those 5 - 12 years and <12 g/dL in those 12 - 15 years.
Investigations
- Complete blood count, including Hb, red cell indices, white cell count, differential count and platelet count.
- Absolute reticulocyte count.
- Serum ferritin level.
- Serum transferrin saturation.
- Serum B12 level.
- Serum folate level.
Management
- Maintain optimal nutrition.
- Exclude other factors that may contribute to anaemia, e.g. infections.
- Current available investigations are unreliable for the monitoring of iron status. Ferritin is an acute-phase reactant that may be elevated in malnutrition and systemic inflammation. The target serum ferritin level in the absence of inflammation is >100 ng/mL or 100 μg/L, and the target transferrin saturation (TSAT) is >20%.
- If iron deficiency is present, treat with an oral iron supplement 6 mg/kg elemental iron/day administered before eating or taking phosphate binders.
- Intravenous iron may be more practical for patients on haemodialysis.
- All children with CKD who are on erythropoietin-stimulating agents (ESA) should be treated with iron (oral or parenteral) to maintain TSAT >20% and a ferritin level >100 ng/mL.
- Consider treatment with ESA when anaemia is persistent despite correction of iron, folate and vitamin B2 deficiencies.
- BP must be controlled before starting ESA treatment.
- Start with 100 U/kg/week subcutaneously in two divided doses.
- Measure Hb, reticulocyte count and ferritin every 4 weeks.
- If the Hb increases <1 g/dL/month, exclude occult infection, blood loss or vitamin B12 or folate deficiency.
- If no cause is found, increase the ESA dose by 25 U/kg/week.
- Continue monitoring the Hb level every 4 weeks and adapt the dose.
- The maximum ESA dose is 300 U/kg/week.
- The target Hb is 10 - 12 g/dL.
- Once the target Hb level of 12 g/dL has been reached, continue with this dose.
- If the Hb increases >12 g/dL, discontinue erythropoietin for one week, and continue with 25 U/kg less per week.
Dyslipidaemia
Dyslipidaemia is an independent risk factor for cardiovascular disease and progression of CKD.
Management
- Prevent malnutrition.
- Treat anaemia, correct metabolic acidosis and manage hyperparathyroidism.
- Lifestyle modification does not have impressive effects in children with dyslipidaemia.
- Consider treatment with a statin (hydroxymethylglutaryl co-enzyme A reductase inhibitor) in children >8 years of age with persistent total cholesterol levels >7 mmol/L.
- Administer simvastatin 10 mg at night (maximum dose 40 mg at night).
- Investigate for rhabdomyolysis if the child complains of muscle cramps.
Fluid and electrolyte balance
Sodium
- Infants with salt-losing CKD (obstructive uropathy and renal hypodysplasia) require NaCl supplementation. A starting dose of NaCl is 1 - 2 mmol/kg/day in divided doses. The dose is guided by serum sodium and chloride levels.
- Salt intake is restricted in patients with oedema, volume overload and HT.
- No salt should be added to food during preparation. Avoid salt-preserved foods.
Water intake is not restricted and should be taken as desired.
Potassium
- Hyperkalaemia often occurs in association with catabolism and metabolic acidosis.
- Limit potassium intake if serum potassium >5.5 mmol/L.
- Restrict potassium-rich foods (fruit juices, fresh fruits and vegetables).
- Soak vegetables for 24 hours before cooking or decant water twice during cooking.
- Discontinue drugs that can cause hyperkalaemia (e.g. ACE inhibitors, β-blockers, spironolactone).
- If serum potassium remains >5.5 mmol/L, treat with sodium polystyrene sulphonate (Kexelate) 1 g/kg/dose dissolved in dextrose water administered orally once or twice daily.
Acid-base status
- Chronic metabolic acidosis causes anorexia and is associated with protein wasting.
- Correction of acidosis improves growth, prevents bone demineralisation and helps to manage hyperkalaemia.
- Treat with NaHCO3- - starting dose 2 mmol/kg/day in 2 - 3 divided doses.
- Maintain serum HCO3- between 18 and 24 mmol/L.
Nutrition and diet
- Protein restriction has no beneficial effects on the progression of CKD in children.
- It is associated with poor growth and the development of malnutrition.
- Children with CKD need a high-calorie and normal protein diet for optimal growth.
- Nasogastric tube feeds/gastrostomy feeds should be given if indicated.
Vitamins and minerals
- Multivitamin 5 mL daily, including vitamins B1, B6, B12 and C.
- Folic acid 2.5 mg daily.
Immunisation
- All children must complete routine immunisations according to the Expanded Programme of Immunisation schedule.
- Pneumovax (23 strain) 0.5 mL intramuscularly is given to children >2 years.
- Varicella zoster virus vaccine, 2 000 pfu/0.5 mL subcutaneously, reconstituted to 3 mL; two doses given with a 6-week dose interval.
- Check immunity against hepatitis B. In the absence of immunity, vaccinate as for any non-immune individual.
- Hepatitis B vaccine 1 mL (3 μg) intramuscular injection, three doses at monthly intervals.
- If the antibody level is considered non-protective or insufficient, give two booster doses one month apart.
Final recommendation
Once a child reaches stage 3 CKD, referral to a paediatric nephrologist is necessary to discuss and/or start planning the potential future need of renal replacement therapy with the parents.
 Correspondence:
Correspondence:
A M Meyers
nkfsa@mweb.co.za

