










Serviços Personalizados
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkSAMJ: South African Medical Journal
versão On-line ISSN 2078-5135
versão impressa ISSN 0256-9574
SAMJ, S. Afr. med. j. vol.104 no.11 Pretoria Nov. 2014
http://dx.doi.org/10.7196/SAMJ.8633
REVIEW
PAEDIATRIC HEPATOBILIARY
Controversies in choledochal malformation
J J AtkinsonI; M DavenportII
IBSc Hons, FRACS (Paeds); Department of Paediatric Surgery, King's College Hospital, London, UK
IIChM, FRCS (Paeds), FRCS (Eng); Department of Paediatric Surgery, King's College Hospital, London, UK
ABSTRACT
Choledochal malformations (some of which are choledochal cysts) may be characterised as an abnormal dilatation of the biliary tract in the absence of acute obstruction. Most appear to be of congenital origin, probably related to distal bile duct stenosis, and almost 15% can now be detected antenatally. Excision and biliary reconstruction using a Roux loop as an open operation is still the standard to compare against, although laparoscopic reconstruction is increasingly reported. This article discusses recent advances in the understanding of choledochal malformation aetiology and classification, together with the role of newer modalites of surgical treatment such as laparoscopic excision and biliary reconstruction. Although these are definitely feasible, care should be taken before dispensing with standard open techniques that have minimal complications and proven long-term benefit.
Choledochal malformations (CMs) may be characterised by inherent dilatation of the biliary tree without evidence of mechanical obstruction. Dilatation varies from florid and cystic to subtle and fusiform, and was first recognised during postmortem dissection by the German anatomist Abraham Vater, who reported his findings in his 1723 thesis.1 The objective of this article is to offer a clinically orientated review and identify and discuss areas of controversy in detail.
Epidemiology
CMs can present at any point in the life cycle from an antenatal scan to the postmortem table, which makes the true incidence hard to define. If biliary atresia is used as a guide for a condition where the incidence is known (1 in 17 000) and the ratio of the two conditions presenting in infancy is taken from a specialist hepatobiliary unit in the UK, then an approximate figure of about 1 in 100 000 live births may be suggested. However, the incidence is much higher in Asian populations, and a female predominance of around 4:1 is consistently found in most series.2-4
There is no obvious hereditary element in most CMs, though a recent review identified 10 pairs of familial occurrence (siblings and offspring) in the literature.5 Conversely, six pairs of monozygotic twins have been described in which a distinct CM appeared in only one pair. Aside from Caroli's syndrome (see below), the condition is usually isolated. Fusiform biliary dilatation occasionally follows the repair of duodenal atresia, although proving this as an acquired aetiology is often difficult.
Classification of choledochal malformations
One early point of controversy is what to call this entity (International Classification of Diseases, 9th revision, clinical modification (ICD-9-CM) diagnosis code 751.69). The older term, choledochal cyst, describes a predominantly spherical or globular biliary dilatation, and is actually only one type of CM. Modern authors have generally adopted a less prescriptive phrase, for example congenital CM (or dilatation).
The original classification was based upon a literature review by Alonso-Lej et al.6 of 94 cases published up until the 1950s and included:
- Type 1: the classic cystic dilatation of the common bile duct
- Type 2: diverticulum from the common bile duct
- Type 3: localised dilatation of the distal common bile duct within the wall of duodenum - the choledochocele.
The Japanese authors Todani et al. developed their own classification during the 1970s and 1980s, defining three variants of type 1, two variants of type 4 (A & B) and adding isolated intrahepatic dilatation as type 5.7 The classification is complex and many authors continue to misquote the original, especially confusing cystic and fusiform.
The King's College Hospital classification (Fig. 1) simplifies the Todani classification into types 1C and 1F (depending on the predominant appearance as cystic or fusiform) and limits type 4 to the combination of intra- and extrahepatic dilatation. This classification has been the basis of our attempts to define pathophysiological characteristics to each type. Other workers have emulated this simpler approach.
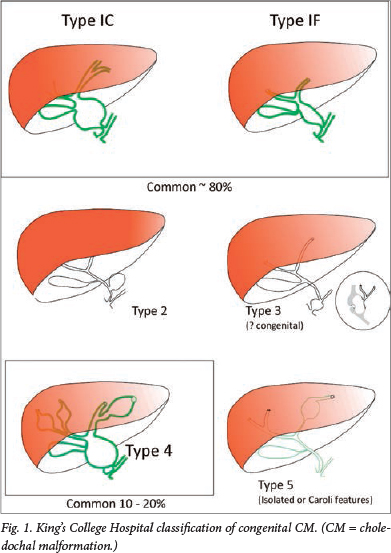
Type 4 CM is found in about 20% of large surgical series of CM and it is arguable whether this is simply the natural history of untreated types 1C and 1F. Some authors have distinguished cystic intrahepatic dilatation from a more fusiform intrahepatic dilatation, implying that the latter diminishes considerably following effective surgery. Our own follow-up study certainly demonstrated considerable resolution in intrahepatic duct dilatation in the first year post surgery.8
Whether the choledochocele (type 3 CM) should be included at all in any classification was the subject of a recent North American series.9 The authors argued that there were so many significant differences when compared with types 1, 2 and 4, particularly age at presentation (invariably as an adult) and gender (predominantly male) that it barely merited the term 'congenital'. Certainly, the distinction between this and a long-standing dilated common channel, coupled with a degree of ampullary stenosis, is difficult.
Isolated type 5 intrahepatic dilatation does occur and is distinct from what we recognise as Caroli's syndrome. This latter entity struggles to fit into any classification of CM because it is so distinctly different. There is an obvious genetic abnormality, leading to a fundamental probable basement membrane defect in small bile ducts. The syndrome is associated with similar fundamental pathology affecting the kidney, manifesting as renal cystic disease or renal fibrosis. Furthermore, there is often associated parenchymal liver fibrosis in the absence of any particular bile duct obstruction.
There is no doubt that part of the CM complex (at least in types 1C, 1F and 4) is a degree of pancreaticobiliary malunion or 'long' common channel, with the junction lying outside of the wall of the duodenum (and therefore not under sphincteric control). Whether it is simply a reflection of disordered bile duct development during fetal life, and hence incidental, or is actually aetiological, as the Babbitt hypothesis implies (see below), remains to be argued.
Aetiology
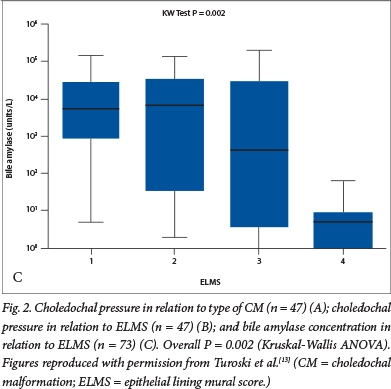
Two hypotheses have been put forward to explain the appearance and configuration of the common types of CM. The most fashionable has been the so-called Babbitt hypothesis,10 which suggests that there is reflux of activated pancreatic enzymes via the common channel into the biliary tract. Donald Babbitt was an American radiologist who first observed this during cholangiography. The hypothesis suggests that this results in enzymatic damage to the local biliary epithelium and weakening of the bile duct wall, leading to dilatation and loss of bile duct structural integrity. An older hypothesis posits that there is a narrow stenotic portion of common bile duct, proximal to the common channel, that generates high intraductal pressures upstream. Prolonged exposure to elevated ductal pressure dilates the proximal lumen, similar to the dilatation seen in the bowel in congenital bowel atresia. Ligation of the duct in fetal lambs by the South African Lewis Spitz certainly produced cystic change in the delivered animals.11 We sought to disentangle these competing hypotheses in a series of reports based on clinical observations at the time of the laparotomy. Initially, we identified an inverse relationship between bile amylase (a surrogate of pancreatic juice reflux) and measured choledochal pressure.12 We also determined that there was a stepwise increase in pressures observed from type 1F through type 1C to type 4 (Fig. 2A), accompanied by a reduction in bile amylases.13 We then related these variables to a semiquantitive histological score, reflecting the damage done to the biliary epithelium, and showed that the most abnormal histology was in bile ducts with the highest pressures (and therefore the lowest amylase levels)(Fig. 2B and 2C).13 These observations in clinical practice seemed to negate the central aetiological mechanism underlying the Babbitt hypothesis.


Clinical features
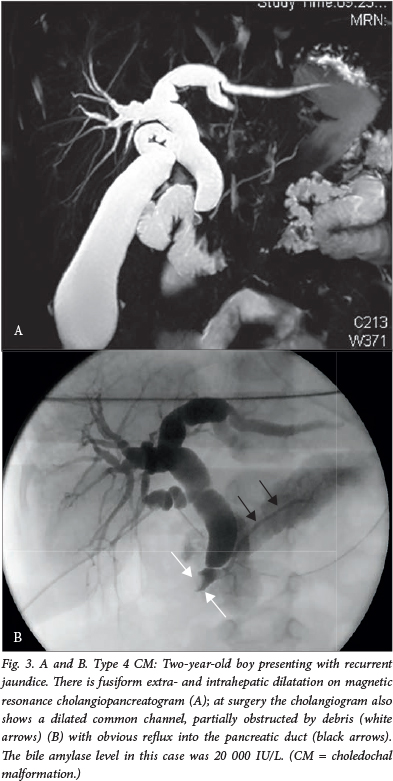
The classic presentation with abdominal pain, jaundice and a mass in the right upper quadrant is in fact rarely seen in large paediatric series.3 Infants tend to present with obstructive jaundice, and in later childhood and adult life, patients present with recurrent abdominal pain, with or without features of acute pancreatitis (Fig. 3).2 , 3 Certainly, recurrent pancreatitis should be seriously considered as a cause for recurrent abdominal pain in childhood and actively excluded by the measurement of serum amylase or lipase. A value of >3 times the upper normal limit, in association with typical abdominal pain and/or features of pancreatitis on cross-sectional imaging (the Atlanta criteria), confirms the diagnosis. Once a diagnosis of pancreatitis has been made, imaging (if not already completed) should at minimum consist of ultrasonography with a low threshold and continue on to a magnetic resonance cholangiopancreatogram and, if necessary, an endoscopic retrograde cholangiopancreatogram. Historically, untreated choledochal cysts caused cirrhosis and death in many instances (mortality rates approached 100% in the 1940s). Prompt surgical resection after diagnosis has rendered this scenario rare in recent times.
Surgical intervention
Early attempts at surgical treatment of choledochal cysts usually involved internal drainage to adjacent intestinal loops or to the duodenum. This approach, leaving the cyst intact, persisted until at least the 1970s.2 The Chicago surgeon G McWhorter is credited with the first successful resection and hepaticoduodenostomy (HD) in 1924.14
Surgery is the only effective treatment for CMs, although the timing can be negotiated. With antenatal detection possible, some obvious type 1C CMs are picked up in the presymptom phase (Fig. 4). Some surgeons advocate immediate surgery during the neonatal period to forestall the development of silent hepatic fibrosis. However, deferment to about 3 - 6 months of age seems more pragmatic as liver biochemistry demonstrably returns to normal after the early neonatal period.

Farello et al.15 first described the laparoscopic resection of a choledochal cyst in a 6-year-old child in 1995. Since then, increasing numbers of specialist centres worldwide have adopted this challenging approach using minimally invasive techniques. In comparison with open surgery, operative times are longer and, certainly in early series, there was a higher incidence of complications such as anastomotic leaks without any real decrease in hospital stay (at least in children). More recently, huge single-institution series of more than 200 cases have been published from Bejing and Hanoi with much lower complication rates. This alludes to the increased centralisation of expertise in those countries and the effect of high-volume surgery.4
Minimally invasive resection is an advanced technique requiring considerable dexterity. Other techniques have been used to try to improve the quality of the biliary anastomosis - the Achilles heel of the entire operation. Thus, robot-assisted surgery has been reported to facilitate manual techniques, although the results from small series show a long learning curve and considerable complications.16 Robots are a prohibitively expensive technology for many healthcare systems, and surgeons in large Asian series do not seem to require them. Extracorporeal jejunojeunostomy through a widened umbilical incision may also reduce operating times. Even single-incision surgery is possible, though apart from marginal improvement in cosmesis, there are no other advantages.
What has been the most controversial recent technical development is a swing back to HD as the reconstruction device. Although the long (40 - 50 cm) Roux loop and hepaticojejunostomy have been the mainstay of biliary reconstructive surgery for approximately the past 50 years, it is difficult to do laparoscopically. An easier alternative is a simple HD. The transected hepatic duct and the mobilised first and second parts of the duodenum can be in close proximity. However, there is a fundamental problem with this technique, which manifested some years ago by surgeons who once preferred it as an open technique.
Todani was a long-time advocate of HD, but also showed that about 10 - 20% of patients have symptomatic bile gastritis due to reflux across the pylorus and a 10% incidence of cholangitis due to reflux of non-sterile duodenal content into the biliary tree, which is often dilated. The final straw, from his perspective, was the development of a bile duct carcinoma at the portal hilum 19 years after primary cyst excision and HD performed on a 13-month-old patient. He then switched to hepaticojejunostomy for the rest of his career. Other authors in comparative trials have also come to the same conclusion. 17 Therefore, what is wrong with hepaticojejunostomy-en-Roux? In our opinion, not much. It is non-physiological and takes a significant amount of absorptive jejunal surface area out of the intestinal sequence. Furthermore, there is constant bile drainage and no intermixing with pancreatic juice until the jejunal union. In a recent study of growth indices in our series of over 100 operated CMs, we found a significant incidence of failure to thrive and lower preoperative weights when compared with the age-corrected normal population. However, after surgery, all patients showed satisfactory catch-up weight gain at 1 year and sustained weight gain thereafter (unpublished observations).
Malignant transformation and cancer risk
The long-term risk of malignant transformation in CM has been well described and is evident in many published series of adults with CM. This risk is probably due to ongoing chronic inflammation in the epithelium over many years, exacerbated by episodic cholangitis and stone formation. The youngest case appears to be an 11-year-old boy, reported by Tanaka et al.18 in 2006, who developed adenocarcinoma in a type 4 CM. Tanaka et al. also identifed ten more cases from the literature in patients aged <20 years, the majority also arising in long-standing type 4 CMs. What is not known with any certainty is the risk of carcinoma in the residual biliary epithelium in the liver or in the head of the pancreas following CM resection in the modern world. We simply have not been doing this kind of surgery long enough to know what happens years later during adulthood. At the very least, however, it should be axiomatic that treated children and adolescents are followed up - ideally by serial ultrasonography. Persistence of intrahepatic dilatation and the development of stones and debris should encourage more invasive investigation.
Discussion of CM and cancer inevitably overlaps with the problems of what to do in patients with an isolated common channel (usually defined as >6 mm long and also referred to in the literature as pancreaticobiliary maljunction). Almost paradoxically, there seems to be a greater malignancy risk with common channels unassociated with CM. In one Japanese survey of hepatobiliary surgical practice, biliary cancer occurred in 22% of adult patients with biliary dilatation and 42% of those without biliary dilatation. Gallbladder cancer is the usual malignancy in the latter group.19
Conclusion
Treatment outcome in CM has improved dramatically over the last 60 years, led by earlier diagnosis, prompt surgical excision and effective biliary drainage. There are still unresolved issues surrounding the nature and style of surgical reconstruction, but long-term success with normal liver function is unequivocal.
REFERENCES
1. Vater A, Ezler CS. Dissertatio de scirrhis viserum occasione sections virii tympanite defunte. Wittenberge: 4 Pamphlers 1723: 881; 22. [ Links ]
2. De Vries JS, de Vries S, Aronson DC, et al. Choledochal cysts: Age of presentation, symptoms, and late complications related to Todani's classification. J Pediatr Surg 2002;37(11):1568-1573. [http://dx.doi.org/10.1053/jpsu.2002.36186] [ Links ]
3. Stringer MD, Dhawan A, Davenport M, Mieli-Vergani G, Mowat AP, Howard ER. Choledochal cysts: Lessons from a 20 year experience. Arch Dis Child 1995;73(6):528-531. [ Links ]
4. Liem NT, Pham HD, Dung LA, Son TN, Vu HM. Early and intermediate outcomes of laparoscopic surgery for choledochal cysts with 400 patients. J Laparoendosc Adv Surg Tech 2012;22(6):599-603. [http://dx.doi.org/10.1089/lap.2012.0018] [ Links ]
5. Iwasaki J, Ogura Y, Nakagawa S, Kato K, Kondo A, Shiraki K. Familial occurrence of congenital bile duct dilatation. World J Gastroenterol 2008;14(6):941-943. [http://dx.doi.org/10.3748/wjg.14.941] [ Links ]
6. Alonso-Lej F, Rever WB Jr, Pessagno DJ. Congenital choledochal cyst, with a report of two and analysis of 94 cases. Int Abstr Surg 1959;108(1):1-30. [ Links ]
7. Todani T, Watanabe Y, Narusue M, Tabuchi K, Okajima K. Congenital bile duct cysts: Classification, operative procedures, and review of thirty-seven cases including cancer arising from choledochal cyst. Am J Surg 1977;134(2):263-269. [ Links ]
8. Hill R, Parsons C, Farrant P, Sellars M, Davenport M. Intrahepatic duct dilatation in type 4 choledochalmalformation: Pressure-related, postoperative resolution. J Pediatr Surg 2011;46(2):299-303. [http://dx.doi.org/10.1016/j.jpedsurg.2010.11.008] [ Links ]
9. Ziegler KM, Pitt HA, Zyromski NJ, et al. Choledochoceles: Are they choledochal cysts? Ann Surg 2010;252(4):683-690. [http://dx.doi.org/10.1097/SLA.0b013e3181f6931f] [ Links ]
10. Babbitt DP. Congenital choledochal cysts: New etiological concept based on anomalous relationships of the common bile duct and pancreatic bulb. Ann Radiol 1969;12(3):231-240. [ Links ]
11. Spitz L. Experimental production of cystic dilatation of the common bile duct in neonatal lambs. J Pediatr Surg 1977;12(1):39-42. [ Links ]
12. Davenport M, Basu R. Under pressure: Choledochal malformation manometry. J Pediatr Surg 2005;40(2):331-335. [http://dx.doi.org/10.1016/j.jpedsurg.2004.10.015] [ Links ]
13. Turowski C, Knisely AS, Davenport M. Role of pressure and pancreatic reflux in the aetiology of choledochal malformation. Br J Surg 2011;98(9):1319-1326. [http://dx.doi.org/10.1002/bjs.7588] [ Links ]
14. McWhorter GL. Congenital cystic dilatation of the common bile duct: Report of a case, with cure. Arch Surg 1924;8(2):604-626. [ Links ]
15. Farello GA, Cerofolini A, Rebonato M, et al. Congenital choledochal cyst: Video-guided laparoscopic treatment. Surg Laparosc Endosc Percutan Tech 1995;5(5):354-358. [ Links ]
16. Dawrant MJ, Najmaldin AS, Alizai NK. Robot-assisted resection of choledochal cysts and hepaticojejunostomy in children less than 10 kg. J Pediatr Surg 2010;45(12):2364-2368. [http://dx.doi.org/10.1016/j.jpedsurg.2010.08.031] [ Links ]
17. Shimotakahara A, Yamataka A, Yanai T, et al. Roux-en-Y hepaticojejunostomy or hepaticoduodenostomy for biliary reconstruction during the surgical treatment of choledochal cyst: Which is better? Pediatr Surg Int 2005;21(1):5-7. [http://dx.doi.org/10.1007/s00383-004-1252-1] [ Links ]
18. Tanaka S, Kubota M, Yagi M, et al. An 11-year-old male patient demonstrating cholangiocarcinoma associated with congenital biliary dilatation. J Pediatr Surg 2006;41(1):e15-e19. [http://dx.doi.org/10.1016/j.jpedsurg.2005.10.066] [ Links ]
19. Kamisawa T, Ando H, Shimada M, et al. Recent advances and problems in the management of pancreaticobiliary maljunction: Feedback from the guidelines committee. J Hepatobiliary Pancreat Sci 2014;21(2):87-92. [http://dx.doi.org/10.1002/jhbp.8] [ Links ]
 Correspondence:
Correspondence:
M Davenport
markdav2@ntlworld.com

