










Serviços Personalizados
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkSAMJ: South African Medical Journal
versão On-line ISSN 2078-5135
versão impressa ISSN 0256-9574
SAMJ, S. Afr. med. j. vol.104 no.11 Pretoria Nov. 2014
http://dx.doi.org/10.7196/SAMJ.8257
RESEARCH
Duchenne muscular dystrophy: High-resolution melting curve analysis as an affordable diagnostic mutation scanning tool in a South African cohort
A I EsterhuizenII; J M WilmshurstIII; R G GoliathIV; L J GreenbergI
IPhD; Division of Human Genetics, Department of Clinical Laboratory Sciences, Institute of Infectious Diseases and Molecular Medicine, Faculty of Health Sciences, University of Cape Town, South Africa
IIMSc (Med); Division of Human Genetics, Department of Clinical Laboratory Sciences, Institute of Infectious Diseases and Molecular Medicine, Faculty of Health Sciences, University of Cape Town, South Africa; National Health Laboratory Service, Groote Schuur Hospital, Cape Town, South Africa
IIIMB BS, MD, MRCP (Lond), FCPaed (SA); Paediatric Neurology and Neurophysiology, Red Cross Children's War Memorial Hospital, School of Child and Adolescent Health, Faculty of Health Sciences, University of Cape Town, South Africa
IVPhD; Division of Human Genetics, Department of Clinical Laboratory Sciences, Institute of Infectious Diseases and Molecular Medicine, Faculty of Health Sciences, University of Cape Town, South Africa; National Health Laboratory Service, Groote Schuur Hospital, Cape Town, South Africa
ABSTRACT
BACKGROUND: Duchenne/Becker muscular dystrophy (D/BMD) is an X-linked recessive muscle disorder affecting 1/3 500 live male births worldwide. Up to 70% of all D/BMD cases are caused by exonic deletions or duplications routinely identified in diagnostic laboratories worldwide. The remaining patients harbour other sequence alterations for which testing availability is limited owing to the expense of interrogating the large DMD gene. Genetic screening for D/BMD in South Africa currently includes multiple ligase-dependent probe amplification (MLPA) for exonic deletions and duplications and linkage analysis. No genetic testing for small mutations in the DMD gene is offered, leaving a third of D/BMD families without genetic closure. The advent of potential mutation-specific therapies for DMD necessitates comprehensive testing protocols
OBJECTIVE: To investigate the effectiveness and affordability of high-resolution melting curve analysis (hrMCA) for detection of small/point mutations in the DMD gene, for possible inclusion into the local public health-funded diagnostic service
METHODS: DNA from 24 patients who had previously tested deletion-negative with multiplex polymerase chain reaction (mPCR) was analysed by MLPA and hrMCA
RESULTS: MLPA revealed eight previously undetected exonic rearrangements: five deletions and three duplications. HrMCA of the remaining samples revealed three nonsense, four frameshifts, one splice-site, one missense and one single-base substitution in the Dp427promoter/exon1 of the DMD gene. In addition, 41 polymorphisms and three changes of uncertain significance were detected
CONCLUSION: These findings identify hrMCA as an affordable and effective mutation scanning tool for incorporation into the local diagnostic setting, allowing for better genetic counselling of more DMD families and selection of potential candidates for future therapies
Duchenne muscular dystrophy (DMD) (OMIM #310200) is the most common of the inherited muscular dystrophies and is caused by lack or faulty production of the dystrophin protein, which is a vital component of the dystrophin-associated protein complex, critical to the maintenance of the structural integrity of muscle fibres. DMD is an X-linked recessive disorder, occurring at an incidence of 1/3 500 live male births worldwide.1 It generally manifests in boys between the ages of 2 and 5 years and is typically marked by delayed motor milestones and symptoms such as frequent falling, difficulty in getting up, gait problems, toe-walking and flat-footedness.2 In untreated patients, respiratory and cardiac complications result in death at a mean age of 19 years. The milder, allelic form of DMD, Becker's muscular dystrophy (BMD), occurs at a lower frequency (1/18 450 live male births), with a later age of onset, a more diverse presentation and a longer life expectancy.3
D/BMD is a monogenic disease, caused by mutations in the dystrophin-encoding DMD gene, which encompasses ~2.5 Mb of genomic sequence and a ~14 kb RNA transcript, comprising 79 exons and eight tissue-specific promoters (Fig. 1). Owing to its large size, the DMD gene is prone to mutations, of which approximately 70% are exonic deletions and duplications. These tend to cluster within two hotspot regions, spanning exons 44 - 53 and 2 - 20. The remaining 25 - 35% of the disease-associated changes are small/point alterations, mostly nonsense, frameshift or splice-site mutations.4 These do not appear to exhibit any clustering effect, and because of the large size of the DMD gene, continue to present a significant diagnostic challenge.

The genetic service for D/BMD at Groote Schuur Hospital, Cape Town, South Africa (SA) and the Division of Human Genetics, University of Cape Town (UCT), commenced in 1987 and was the first of its kind to be offered nationally by the state health services in SA.5 The early testing protocol involved tracking the inheritance of the putative X chromosome using intragenic linkage markers and later included the deletion hotspot screen with multiplex polymerase chain reaction (mPCR), based on the work published by Chamberlain et al.6 In 2000, the Human Genetics Laboratory in Johannesburg (now also incorporated into the National Health Laboratory Service (NHLS)) began diagnostic testing for D/BMD, using a similar protocol of mPCR and linkage analysis.7 The genetic service for D/BMD, currently offered by both centres, is extended to state and private patients in and outside SA.
Introduction of the multiple ligase-dependent probe amplification (MLPA) in 2007 by the NHLS in Cape Town and Johannesburg significantly improved the level of service, as the detection range of the assay spans all exons of theDMD gene, allowing for identification of exonic rearrangements across the gene, not just the hotspots. Furthermore, the dosage component of the MLPA enables identification of duplications in males and determination of female carrier status, which could not be achieved with the mPCR.8 Determining the female carrier status is a significant aspect of any molecular genetic service for D/BMD, not only for the purpose of effective genetic counselling but also for clinical monitoring and management of D/BMD carriers, who develop symptoms in approximately 22% of all cases. These may involve a cardiac pathology (dilated cardiomyopathy) with or without muscle weakness, which varies from very mild to a DMD-like clinical course.9 Another advantage of the MLPA analysis for D/BMD is better delineation of the extent of the exonic deletion/duplication in an individual.
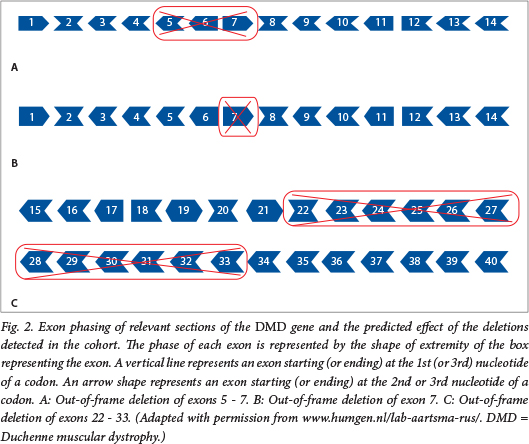
Genotype-phenotype correlation studies in D/BMD have shown that the disease severity is influenced not so much by the size of the mutation, as by: (i) the location of the mutation in relation to the critical functional domains of the dystrophin protein; and (ii) the effect of the mutation on the DNA translational open reading frame (ORF).10 , 11 This so-called 'reading-frame hypothesis' argues that mutations that disrupt the ORF result in more severe phenotypes (DMD) than those where the reading frame is retained (BMD). The effect of an exonic rearrangement on the ORF and possibly the phenotype can be predicted, as the intron-exon boundaries have been well characterised and show that some exons of the DMD gene do not contain an integral number of triplet codons and if deleted will result in a frameshift of the mRNA (Fig. 2). While such predictions made on DNA-based test results should be approached with caution, they are thought to add value, especially in early diagnoses of cases with no family history, where possible anticipation of the disease severity may influence the approach to treatment. Nevertheless, molecular confirmation of the clinical diagnosis of B/DMD remains elusive to approximately a third of the SA D/BMD patient population, i.e. those carrying small/point mutations.
Recent advances in the development of mutation-specific therapies for DMD support routine availability of testing for most D/BMD-causing mutations. While next generation sequencing and microarray studies present the ultimate technological approach and have been successfully employed by investigators abroad,12 , 13 the platforms are prohibitively expensive in the local public health context and the bioinformatic support structure required for routine implementation is largely unavailable at present, though much effort is being made in that direction. Similarly, while the cost of direct BigDye Terminator sequencing (Life Technologies, USA) is no longer seen as high in the developed countries, it is still an expensive option in SA, especially considering the amount of sequencing required for analysis of the vast DMD gene. The work presented here was undertaken in response to the call from the clinical community in SA for local availability of affordable testing for all, or most, B/DMD-causing mutations.
Objective
The project was conducted as a pilot study aimed at investigating the effectiveness and affordability of high-resolution melting curve analysis (hrMCA) as a presequencing scanning tool for detection of small/point mutations in the DMDgene, for possible inclusion into the local public health-funded diagnostic service.
Methods
Patients and controls
The study panel included 24 unrelated boys diagnosed with D/BMD on the basis of clinical presentation and elevated creatine kinase levels. Laboratory confirmation of the diagnosis by immunohistochemistry (IHC) of muscle biopsy samples was available in 13 cases. IHC results were not available or muscle biopsies were not performed on the remaining 11 patients. Of the 24 boys, all of whom had previously tested deletion-negative with the mPCR hotspot screen offered routinely before 2007, 19 had been referred from the Neuromuscular Clinic at Red Cross War Memorial Children's Hospital in Cape Town and five from other centres in the country. The cohort ethnicity reflected the patient base of the referral centres. Where available, archived DNA was retrieved from the DNA Registry (REC/REF 234/2010) at the Division of Human Genetics, UCT. Alternatively, DNA was extracted from blood lymphocytes, using standard techniques. DNA samples from two healthy male volunteers were obtained for inclusion as wild-type controls. The study protocol was approved by the Human Research Ethics Committee, UCT (REC/REF 416/2008), in keeping with the tenets of the Declaration of Helsinki (2008).
MLPA analysis
Owing to the limitations of the mPCR method, all samples were retested for exonic rearrangements using the MLPA (PO34 and P035 probe mixes (MRC-Holland, Netherlands). The MLPA products were separated by capillary electrophoresis on the ABI 3130xl Genetic Analyser (Life Technologies, USA) and the data files analysed with the GeneMapper v4.1 (Applied Biosystems, USA) and the Coffalyser.NET software (www.mlpa.com) (details available on request). The probable effect of the exonic changes on the ORF was predicted using the Reading Frame Checker (http://www.humgen.nl/scripts/DMD_frame.php).
Mutation scanning with hrMCA
Principle
HrMCA is based on the natural process of DNA denaturation or 'melting' upon exposure to a gradual increase in temperature. Double-stranded DNA (dsDNA) dissociates into single-stranded DNA (ssDNA) in the presence of an intercalating dye, which fluoresces only while incorporated into dsDNA. The melting process can be monitored by measuring the gradually diminishing amount of fluorescence during strand dissociation, and a melting curve is derived by plotting fluorescence v. temperature (Fig. 3). It has been recognised that the precise shape of the melting curve is a function of the DNA sequence. The observed thermal denaturation profile is characteristic of a specific DNA fragment and is dependent on its sequence length, base and guanine-cytosine (GC) content.14 Successful hrMCA requires specialised instrumentation capable of collecting data with exquisite real-time and thermo-optical precision. In this study, DNA of patients who tested MLPA-negative for exonic rearrangements was subjected to hrMCA on the RotorGene™6000 (Corbett Research, Australia), which is a real-time PCR and hrMCA platform suitable for a reliable high-resolution melting (HRM) analysis.15

PCR amplification and hrMCA
The entire coding region of the DMD gene in each patient was PCR-amplified and subjected to hrMCA on the RotorGene™600, using 96 sets M13-tailed primer pairs and the EvaGreen (Biotum Inc.) intercalating dye. The primers were specifically designed for hrMCA and subsequent sequencing as published by Almomani et al.16 Amplification efficiency in real time and hrMCA profiles of the test v. the wild-type DNA samples were compared by visual scrutiny and RotorGene™6000 Series Analysis Software (details available on request).
Sequencing
Fragments exhibiting altered HRM profiles were sequenced with the universal M13 sequencing primers and the BigDye®Terminator v3.1 mix (Life Technologies, USA), using standard methods. The sequences were compared to the Leiden coding reference sequence (www.dmd.nl), based on the GenBank reference file NM_004006.1 of the Dp427m dystrophin isoform (with one difference: 12505G>A). The output files were analysed with the BioEdit Sequence Alignment Editor and the NCBI Basic Local Alignment Search Tool (BLAST) (www.ncbi.nlm.nih.gov).
Bioinformatic analysis
Possible disease association of the sequence variants was determined in silico using the ESEfinder 2.0, Human Splicing Finder (HSF), Sorting of Intolerant From Tolerant (SIFT), Transcription Element Search System (TESS). It must be emphasised that the results of such analyses are indicators of probable effects rather than definitive proof thereof. The final effect of a given mutation may be influenced by a combination of factors not necessarily taken into account during a computational search for a specific effect.
Results
MLPA analysis
The MLPA revealed eight exonic changes (five deletions and three duplications) previously undetected with the mPCR assay (Table 1), which was not capable of detecting duplications or deletions located outside of its range, largely limited to the deletion hotspots. The superior informativity of the MLPA in comparison with the previously used mPCR was clearly demonstrated here (as also shown by colleagues in Johannesburg).7 The MLPA findings were confirmed by repeat MLPA and single-exon PCRs. No further analyses were carried out to confirm pathogenicity, as exonic rearrangements are an accepted cause of D/BMD.
Interestingly, the predicted effects of both duplication mutations detected were in conflict with the recorded phenotype. This is in fact consistent with the literature, where authors warn that while reading-frame predictions are useful, DNA-based predictions of duplications in particular should be viewed with caution, as the true nature of the rearrangements may not be known owing to events such as non-tandem rearrangements or altered splicing.10
HrMCA and sequencing
At least one hrMCA variant profile was noted in each of the 16 patients screened and variant profiles were noted in 218 (14%) of all the analysed PCR fragments (96 per patient). Cycle sequencing revealed sequence alterations in 54 (3%) of these hrMCA variants. This high number of false variants was attributed to the variable quality of the available DNA, as fresh blood specimens could not be obtained in every case. Also, the criteria applied for variant selection erred on the side of caution, to avoid missing disease-associated mutations.
Ten of the sequence alterations found were classified as disease-causing (Table 2) and comprised seven truncating mutations (three nonsense and four frameshifts), one splice-site, one missense and one point substitution in the Dp427 promoter/exon1 region of the DMD gene. Of these, only two (c.4729C>T and c.6905G>A) (Fig. 3) had previously been recorded in the Leiden Open Variation Database (LOVD), Leiden muscular dystrophy pages (http://www.dmd.nl). 4 The remaining eight mutations were accepted as novel, based on the LOVD and a literature search. Truncating mutations (nonsense and frameshifts) in the DMD gene are generally accepted as pathogenic, as are mutations affecting splicing (Table 2). Bioinformatics tools (previously listed) were used to confirm this, and to determine the probable pathogenicity of the novel missense mutation in exon 7 and the 5ʹUTR sequence change (Table 2). Testing of population cohorts of affected and wild-type samples yielded negative results, providing further evidence towards the pathogenic and private (i.e. patient/family specific) nature of both these mutations. It must again be mentioned that while the disease association suggested by such analyses is likely, a definitive elucidation of their role in disease pathogenesis can only be achieved with mRNA expression and functional studies, all of which were beyond the scope of this project.
Where possible, archived DNA from family members was tested with PCR, restriction endonuclease (RE) or sequence analysis for the putative change identified in the proband. In each case, the mutation was shown to segregate with the affected phenotype. This did not provide definitive proof as the variant could simply segregate with the inherited chromosome, but went towards strengthening the case for pathogenicity.
Also identified were 41 benign polymorphisms, two of which were novel (data not shown). The assumption of non-pathogenicity was based their presence in multiple patients, co-segregation with pathogenic mutations, and/or previous reports of non-pathogenicity in the LOVD. Study findings also revealed three sequence alterations of uncertain clinical significance (data not shown), viewed as probably benign owing to their location at the 3ʹUTR and close proximity to other changes listed as non-pathogenic in the DMD LOVD. However, since no other pathogenic changes were found in these patients, additional studies are required to definitively exclude or establish their significance.
No disease-associated mutations were identified in eight patients in the cohort. The reasons for this may be threefold. Firstly, the clinical diagnosis was not confirmed with muscle biopsies in four of these cases and in the absence of genetic diagnoses, clinical reassessments may be warranted. Secondly, in some cases only archived DNA extracted using different methods was available, which may have resulted in missed variants owing to compromised DNA integrity or the presence of impurities known to influence the outcomes of hrMCA. Lastly, some of the patients could carry deep intronic mutations resulting in novel splice sites and inclusion of intronic sequences into the mRNA. These 'pseudoexon mutations' cannot be picked up with DNA-based testing and the methods used here, owing to the exceptionally large size of the DMD introns, and require analysis of mRNA from muscle tissue.17
Discussion
In recent years, much effort has gone into investigating methodologies for comprehensive mutation detection in D/BMD, driven by the prospects of availability of mutation-based therapies. With that in mind, the major aim of this study was testing hrMCA as a screening tool for detection and characterisation of small/point mutations in the DMDgene in the local laboratory environment. We view identification of pathogenic mutations in ten out of 16 patients tested as a positive outcome, which has provided a significant part of the cohort and their families with previously unavailable genetic diagnoses.
The method employed (hrMCA) was chosen for its ease and cost-effectiveness as a mutation scanning tool. In contrast to the diagnostic laboratories in developed countries, where DNA sequencing is relatively inexpensive, the local cost of direct sequencing is high, at approximately ZAR100.00 per PCR fragment at the time of the study. This is a conservative estimate, which only partially factors in the cost of consumables and the analyst's time. The size of theDMD gene, where approximately 96 PCR fragments must be sequenced to cover its coding region (with genomic DNA as a template), makes direct sequencing in the local context prohibitively expensive. This cost is considerably reduced with the use of a presequencing mutation scanning tool such as the hrMCA, at ~R7.00 per PCR fragment. Once a putative 'family' mutation is identified in the proband, testing of other members of the family can be performed cheaply by targeting a specific region of the gene with PCR, RE or sequencing analysis.
Research involving point mutation detection in the DMD gene has previously been conducted locally. However, this is the first study in SA, and to our knowledge on the African continent, to take an all-encompassing approach to mutation profiling across the coding region of the DMD gene, i.e. detection of exonic rearrangements with the MLPA and small/point mutation detection with hrMCA and sequencing. Future plans for the molecular genetic service for DMD include establishing massive parallel sequencing as a diagnostic platform, which will further improve local mutation detection capabilities. An additional consideration is the superior nature of RNA-based analysis for definitive determination of the downstream effects of sequence alterations.16 However, this requires muscle tissue sampling, which is invasive and is typically undertaken only in cases of clinically suspected D/BMD with negative results of molecular studies. This may change in the future, should RNA analysis become essential for ascertainment of eligibility for a mutation-based therapy. Until such time, however, DNA extracted from blood lymphocytes is likely to remain the mainstay of diagnostic testing.
Conclusion
The work described here provides the foundation for establishing a cost-effective, comprehensive and specific testing strategy for every D/BMD family. Furthermore, defining the mutations underlying the D/BMD phenotype in the SA population will facilitate establishing trends within the various population subgroups of SA, possibly correlating molecular features to the disease course and facilitating a more strategised approach to the clinical and laboratory diagnostic protocols. The knowledge of the disease-causing mutations detected during this study provides patients and their families with a considerable head-start in the future, when mutation-based genetic therapy becomes a veritable therapeutic avenue.
Acknowledgments.The study was funded by the Muscular Dystrophy Foundation, SA. The authors wish to thank the Foundation for their enthusiasm and support throughout.
REFERENCES
1. Emery AE. The muscular dystrophies. Lancet 2002;359(9307):687-695. [http://dx.doi.org/10.1016/S0140-6736(02)07815-7] [ Links ]
2. Bushby K, Finkel R, Birnkrant DJ, et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: Diagnosis, and pharmacological and psychosocial management. Lancet Neurol 2010;9(1):77-93. [http://dx.doi.org/10.1016/S1474-4422(09)70271-6] [ Links ]
3. Blake DJ, Kroger S. The neurobiology of Duchenne muscular dystrophy: Learning lessons from muscle? Trends Neurosci 2000;23(3):92-99. [http://dx.doi.org/10.1016/S0166-2236(99)01510-6] [ Links ]
4. Aartsma-Rus A, van Deutekom JC, Fokkema IF, van Ommen GJ, den Dunnen JT. Entries in the Leiden Duchenne muscular dystrophy mutation database: An overview of mutation types and paradoxical cases that confirm the reading-frame rule. Muscle Nerve 2006;34(2):135-144. [http://dx.doi.org/10.1002/mus.,20586] [ Links ]
5. Ballo R, Viljoen D, Beighton P. Duchenne and Becker muscular dystrophy prevalence in South Africa and molecular findings in 128 persons affected. S Afr Med J 1994;84(8):494-497. [ Links ]
6. Chamberlain JS, Gibbs RA, Ranier JE, Nguyen PN, Caskey CT. Deletion screening of the Duchenne muscular dystrophy locus via multiplex DNA amplification. Nucleic Acids Res 1988;16(23):11141-11156. [http://dx.doi.org/10.1093/nar/16.23.11141] [ Links ]
7. Kerr R, Robinson C, Essop FB, Krause A. Genetic testing for Duchenne/Becker muscular dystrophy in Johannesburg, South Africa. S Afr Med J 2013;103(12):999-1004. [http://dx.doi.org/10.7196/samj.7274] [ Links ]
8. Gatta V, Scarciolla O, Gaspari AR, et al. Identification of deletions and duplications of the DMD gene in affected males and carrier females by multiple ligation probe amplification (MLPA). Hum Genet 2005;117(1):92-98. [http://dx.doi.org/10.1007/s00439-005-1270-7] [ Links ]
9. Brioschi S, Gualandi F, Scotton C, et al. Genetic characterization in symptomatic female DMD carriers: Lack of relationship between X-inactivation, transcriptional DMD allele balancing and phenotype. BMC Med Genet 2012;13(1):73. [http://dx.doi.org/10.1186/1471-2350-13-73] [ Links ]
10. Kesari A, Pirra LN, Bremadesam L, et al. Integrated DNA, cDNA, and protein studies in Becker muscular dystrophy show high exception to the reading frame rule. Hum Mutat 2008;29(5):728-737. [http://dx.doi.org/10.1002/humu.20722] [ Links ]
11. Muntoni F, Torelli S, Ferlini A. Dystrophin and mutations: One gene, several proteins, multiple phenotypes. Lancet Neurol 2003;2(12):731-740. [http://dx.doi.org/10.1016/S1474-4422(03)00585-4] [ Links ]
12. Ishmukhametova A, van Kien PK, Méchin D, et al. Comprehensive oligonucleotide array-comparative genomic hybridization analysis: New insights into the molecular pathology of the DMD gene. Eur J Hum Genet 2012;20(10):1096-1100. [http://dx.doi.org/10.1038/ejhg.2012.51] [ Links ]
13. Vasli N, Bohm J, Le Gras S, et al. Next generation sequencing for molecular diagnosis of neuromuscular diseases. Acta Neuropathol 2012;124(2):273-283. [http://dx.doi.org/10.1007/s00401-012-0982-8] [ Links ]
14. Erali M, Voelkerding KV, Wittwer CT. High resolution melting applications for clinical laboratory medicine. Exp Mol Pathol 2008;85(1):50-58. [http://dx.doi.org/10.1016/j.yexmp.2008.03.012] [ Links ]
15. White H, Potts G. Mutation Scanning by High Resolution Melt Analysis. Evaluation of Rotor-Gene 6000 (Corbett Life Science), HR-1 and 384-well LightScanner (Idaho Technology). Wessex: National Genetics Reference Laboratory, 2006. [ Links ]
16. Almomani R, van der Stoep N, Bakker E, den Dunnen JT, Breuning MH, Ginjaar IB. Rapid and cost effective detection of small mutations in the DMD gene by high resolution melting curve analysis. Neuromuscul Disord 2009;19(6):383-390. [http://dx.doi.org/10.1016/j.nmd.2009.03.004] [ Links ]
17. Gurvich OL, Tuohy TM, Howard MT, et al. DMD pseudoexon mutations: Splicing efficiency, phenotype, and potential therapy. Ann Neurol 2008;63(1):81-89. [http://dx.doi.org/10.1002/ana.21290] [ Links ]
 Correspondence:
Correspondence:
A I Esterhuizen
alina.esterhuizen@nhls.ac.za
Accepted 26 June 2014.


{kind=link}
{kind=link}