










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSAMJ: South African Medical Journal
On-line version ISSN 2078-5135
Print version ISSN 0256-9574
SAMJ, S. Afr. med. j. vol.104 n.10 Pretoria Oct. 2014
FORUM
CLINICAL ALERT
The influence of glucocorticoids on lipid and lipoprotein metabolism and atherosclerosis
I L RossI; A D MaraisII
IDr Ian Ross is a senior consultant endocrinologist attached to the Division of Endocrinology, Department of Medicine, Faculty of Health Sciences, University of Cape Town, South Africa, and Groote Schuur Hospital, Cape Town. His major interests are Addison's disease, glucocorticoids, clinical thyroidology and thyroid cancer. He received his PhD from UCT on the clinical aspects of Addison's disease
IIProf. David Marais specialised in internal medicine, then received a Medical Research Council scholarship to study lipoprotein metabolism in medical biochemistry before resuming a consultant post in internal medicine at Groote Schuur Hospital. He joined the lipid clinic in 1984 and developed a diagnostic and research laboratory for dyslipidaemias. He headed the clinic from 1990 to 2012, when he joined the Department of Chemical Pathology, National Health Laboratory Service and Faculty of Health Sciences, UCT. He is also a member of the MRC Cape Heart Group
ABSTRACT
Glucocorticoids have multiple therapeutic uses, but their impact on lipid metabolism and cardiovascular disease risk is not always considered during long-term treatment. Genetic variations, environmental factors and the reasons for glucocorticoid treatment all influence the lipid profile and atherosclerosis. Responses to glucocorticoid treatment may therefore be variable and unpredictable. Despite the frequency with which pharmacological doses of glucocorticoids are used, surprisingly few publications examine their effects on lipid metabolism and atherosclerosis. Patients managed with glucocorticoids should have their cardiovascular risk assessed, especially if long-term treatment is planned. While some apparent favourable changes have been reported in high-density lipoprotein metabolism, very-low-density lipoprotein and low-density lipoprotein responses seem unfavourable. The impact of glucocorticoids on atherosclerosis, which is often viewed as an inflammatory process, is unclear. Glucocorticoid treatment should be undertaken for appropriate indications, but in some instances special attention should be given to management of dyslipidaemia, as long-term survivors of treatment are likely to encounter atherosclerosis.
Lipid transport
Lipoproteins transport lipids in the circulation in four major pathways: (i) a postprandial (exogenous) pathway for chylomicrons; (ii) an endogenous pathway involving very-low-density lipoprotein (VLDL) for triglyceride (TG) transport from the liver; (iii) a low-density lipoprotein (LDL) pathway from a proportion of VLDL as a source of cholesterol for cells; and (iv) a reverse cholesterol transport pathway by high-density lipoprotein (HDL).[1] These pathways and the reported effects of glucocorticoids are shown in Fig. 1.
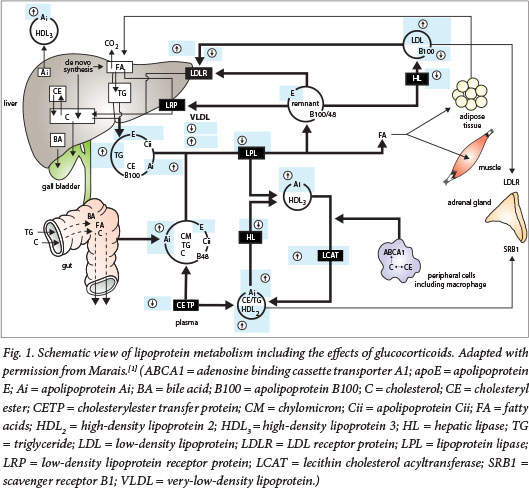
Exogenous TG pathway
Chylomicrons, comprising 85 - 90% TG and containing apolipo-protein B (apo B)-48 (apoB48), apolipoprotein Ai (apoAi) and apolipoprotein Aiv (apoAiv), are produced in enterocytes, traverse the thoracic duct and ultimately reach the systemic circulation. Lipoprotein lipase anchored on cells by heparan sulphate proteoglycans hydrolyses TG at the vascular endothelium, yielding non-esterified fatty acids (NEFAs) and remnants, proportionately richer in cholesterol esters. Chylomicron remnants are rapidly cleared by liver remnant receptors,[2] as a result of apolipoprotein E (apoE) acquired in the circulation. Dietary fat restriction will have a significant impact on severe hypertriglyceridaemia.
Endogenous TG pathway
VLDL is assembled on apolipoprotein B-100 (apoB100) and comprises 50% TG, 20% cholesterol esters, 15% phospholipids and 15% protein. Secretion is enhanced by increasing delivery of NEFAs from adipose tissue during starvation or in diabetes.[3] VLDL is also hydrolysed by lipoprotein lipase. These remnants and other small lipoproteins (LDL and HDL) can undergo hydrolysis of TG by hepatic lipase, forming progressively smaller particles. VLDL remnants are proportionately richer in cholesterol, and some form LDL.[1] The release of fatty acids from adipose tissue and their uptake in the liver will enhance VLDL production and may cause hypertriglyceridaemia.
LDL pathway
LDL contains the majority of cholesterol in the plasma. Its mass comprises 35% cholesteryl ester, 10% unesterified cholesterol (UC), 10% TG and 20% phospholipids. ApoB100 almost entirely accounts for the 25% of protein. Most circulating LDL is taken up by hepatocyte LDL receptors. Increased VLDL could increase LDLC while also resulting in modulation of particle size. This process requires cholesterylester transfer protein (CETP) to enrich with TG, after which hepatic lipase hydrolyses the TG. The plasma LDL concentration may also be raised by decreased clearance (by LDL receptors) in familial hypercholesterolaemia.
Reverse cholesterol transport
HDL is the smallest of the lipoproteins. About half is lipids (25% phospholipids and 15% cholesterylester, while UC and TG both constitute 5%). The remainder is chiefly apoAi and apolipoprotein Aii (apoAii). The liver and intestine secrete apoAi that may initiate particle formation, which may also result from lipolysis of TG-rich lipoproteins[4] when apoAi and the relative excess of phospholipids pinch off from the lipoprotein. Lecithin-cholesterol acyltransferase (LCAT) esterifies UC, using long-chain fatty acids from phospholipids. Cholesteryl esters migrate to the core, forming more mature spherical particles (HDL3) and later larger and less dense HDL. CETP transfers cholesteryl ester from HDL2 to TG-rich lipoproteins, permitting delivery of cholesterol to the liver, and in exchange HDL receives TG.[4] Hepatic lipase hydrolyses TG, regenerating smaller HDL3 particles. Exchange of TG into LDL similarly produces smaller particles. In HDL, esterification of UC permits more UC to be accepted from cells or other lipoproteins. HDL delivers cholesterol directly to the liver, leading to its excretion in bile.[5]
Lipid and lipoprotein changes with corticosteroids
Dyslipidaemia, hyperglycaemia and hypertension are the most significant cardiovascular adverse effects resulting from glucocorticoid therapy,[6] but mechanistic insights are incomplete. Documented changes in human lipid profiles on varying doses of prednisone[7-10] include elevated VLDL, TG and LDL cholesterol, and either increased or decreased HDL cholesterol.
Animal studies of lipid changes in steroid use
Hydrocortisone (single dose) administered to rabbits with atherosclerosis raised TG but not total cholesterol (TC),[11] suggesting increased VLDL production or possibly decreased metabolism. In rats, dexa-methasone and triamcinolone (but not hydrocortisone) increased plasma TC and TG.[12] Hydrocortisone administered to rats at 100 µg/g of body mass reduced TC. Hydrocortisone, triamcinolone and dexamethasone increased apoAi, with the greatest increases documented for triamcinolone and dexamethasone. Dexametha-sone raised apoAiv the most, and triamcinolone caused the greatest increase in apoE, yet reductions in apoE levels occurred in rats receiving hydrocortisone.[12] Methyl-prednisone administered to normal rats for 8 days increased TG and almost doubled TC,[13] probably owing to a reduction in lipoprotein lipase activity and decreased HDL cholesterol.[14] ApoE decreased with hydrocortisone, either as a result of less hepatic secret ion or increased catabolism of apoE-containing lipoproteins, but lower production of apoE by extra-hepatic tissues has also been proposed.[15] The brain, spleen and kidney produce apoE, aiding redistribution of cholesterol from cells with an excess of cholesterol to those requiring it.[15] ApoAi increased with most glucocorticoids, but especially with triamcinolone and dexamethasone, resulting in increased HDL cholesterol.[12,15] Hepatic apoAi mRNA increased in cultured rat hepatocytes exposed to glucocorticoids.[16] Transient down-regu-lation of LDL receptors in rats followed methylprednisolone administration, accounting for elevated LDL and TC.[17] Overall, animal models illustrate marked effects on HDL and some adverse effects on LDL, as well as differences between the drugs.
Human studies with glucocorticoids
The impact of glucocorticoid hormones on lipoprotein metabolism can be examined in normal variation, acute and chronic dosing, replacement therapy, and hypercortisolism. Positive correlations exist between LDL cholesterol and endogenous plasma cortisol in healthy men aged between 52 years and 67 years.[18] Glucocorticoids alter plasma lipids within 14 days.[10] Acute effects of 3 mg dexamethasone (twice daily simulating acute stress) in young men included lower highly sensitive C-reactive protein levels and increased HDL cholesterol; LDL cholesterol, NEFA and TG were not altered.[19] Glucocorticoids reduce hepatic lipase and CETP, resulting in elevated HDL cholesterol after cardiac transplantation.[20] In the third National Health and Nutrition Examination Survey, glucocorticoid use was associated with higher HDL and lower TC/HDL cholesterol ratios.[21] Both glucocorticoid use and endogenous hypercortisolism (Cushing's disease) resulted in elevated TC and LDL cholesterol. Glucocorticoid replacement in hypopituitary patients lowered VLDL, LDL cholesterol, LCAT and CETP.
Based on animal and human studies, exposure to glucocorticoids may produce either increased or decreased HDL cholesterol. Changes in reverse cholesterol transport or other effects may modulate atherosclerosis. Some studies corroborate up-regulated hepatic LDL receptor activity, explaining a decrease in LDL cholesterol. While glucocorticoids are known to have pleiotropic actions on physiological and pathological processes, lipoprotein responses and homoeostasis are varied, but are potentially atherogenic (Table 1).
Hypercortisolism stimulates the production of VLDL.[6] Subclinical Cushing's syndrome has been associated with dyslipidaemia. Rheumatoid arthritis sufferers frequently have high TC and LDL cholesterol and decreased HDL cholesterol. Untreated rheumatoid arthritis patients may have lower HDL cholesterol levels relating to inflammation and acute-phase response. Treatment with glucocorticoids may dampen inflammation favourably, though this may not apply to atherogenesis.[71 A meta-analysis found an increase of cardiovascular and cerebrovascular disease by 59% and 50%, respectively, compared with the general population. Accelerated atherosclerosis in systemic lupus erythematosus has been attributed to the disease or to glucocorticoid therapy.
Hypopituitary patients on replacement therapy (hydrocortisone, thyroxine and sex steroids) are subject to increased morbidity and mortality from accelerated atherosclerosis. Optimally replaced patients had adverse lipid profiles, with increased TG, TC and LDL cholesterol compared with controls. Daily hydrocortisone supplementation of less than 20 mg/d in growth hormone-replaced patients had the least metabolic consequences.
Clinical approach to glucocorticoid treatment
Doctors considering glucocorticoid treatment in patients with chronic disorders should be aware that cardiovascular risk may increase. Chronic inflammatory conditions can predispose to vascular disease, and treatment may aggravate risk through dyslipoproteinaemia or other mechanisms. Until further studies inform otherwise, prevailing guidelines should be followed. Risk calculations based on clinical parameters and lipid profiles as suggested guidelines offer the best guidance on the threshold for treatment, but may not be accurate. The premorbid lipid profile as well as levels during the illness may guide management. Exercise and dietary recommendations should be the norm.
Detailed clinical assessments of a personal and family history of premature cardiovascular disease, physical signs and lipoprotein profiles will assist in the diagnoses listed in Table 2. Physical signs are not invariably present. Certain recessive disorders, e.g. dysbetalipoproteinaemia in subjects homozygous for apolipoprotein E2 (apoE2), manifest only when metabolic stress occurs. Partial lipoprotein lipase activity in heterozygotes may predispose to hypertriglyceridaemia. It is expected that glucocorticoid therapy will have a small impact on the lipoprotein profile in patients with normal genetic constitutions, while benefiting the chronic inflammatory condition. Occasionally, severe dyslipidaemia may be precipitated by glucocorticoid treatment, and in this setting special treatment with statins will be required for LDL hypercholesterolaemia, or fibrates for severe hypertriglyceridaemia. Successful treatment of the nephrotic syndrome with glucocorticoids will result in improved lipid profiles. Precipitation of diabetes by glucocorticoid therapy can affect the lipid profile and cardiovascular risk. Hypertension will similarly require a re-evaluation of risk and preventive actions to combat cardiovascular disease.

Conclusions
Treatment of conditions requiring glucocorticoids together with disease-modifying agents is likely to prolong life expectancy and therefore raise the risk of cardiovascular disease. This risk is related at least in part to lipoprotein responses, as summarised in this article. More studies are required to evaluate cardiovascular risk in replacement and anti-inflammatory treatment, as well as the effects of different doses and forms of corticosteroid.
References. For a complete set of references, please contact the corresponding author.
1. Marais AD. Lipids, lipoprotein metabolism and their derangements. SA Heart 2005;2(3):8-18. [ Links ]
2. Mahley RW, Huang Y, Rall SC Jr. Pathogenesis of type III hyperlipoproteinemia (dysbetalipoproteinemia): Questions, quandaries, and paradoxes. J Lipid Res 1999;40(11):1933-1949. [ Links ]
3. Adiels M, Olofsson SO, Taskinen MR, Boren J. Overproduction of very low-density lipoproteins is the hallmark of the dyslipidemia in the metabolic syndrome. Arterioscler Thromb Vasc Biol 2008;28(7):1225-1236. [http://dx.doi.org/10.1161/ATVBAHA.107.160192] [ Links ]
4. Davidson MH, Toth PP. High-density lipoprotein metabolism: potential therapeutic targets. Am J Cardiol 2007;100(11A):32-40. [http://dx.doi.org/10.1016/j.amjcard.2007.08.011] [ Links ]
5. Rader DJ, Alexander ET, Weibel GL, Billheimer J, Rothblat GH. The role of reverse cholesterol transport in animals and humans and relationship to atherosclerosis. J Lipid Res 2009;50(Suppl):S189-S194. [http://dx.doi.org/10.1194/jlr.R800088-JLR200] [ Links ]
6. McDonough AK, Curtis JR, Saag KG. The epidemiology of glucocorticoid-associated adverse events. Curr Opin Rheumatol 2008;20(2):131-137. [http://dx.doi.org/10.1097/BOR.0b013e3282f51031] [ Links ]
7. Garcia-Gomez C, Nolla JM, Valverde J, Narvaez J, Corbella E, Pinto X. High HDL-cholesterol in women with rheumatoid arthritis on low-dose glucocorticoid therapy. Eur J Clin Invest 2008;38(9):686-692. [http://dx.doi.org/10.1111/j.1365-2362.2008.01994.x] [ Links ]
8. Becker DM, Chamberlain B, Swank R, et al. Relationship between corticosteroid exposure and plasma lipid levels in heart transplant recipients. Am J Med 1988;85(5):632-638. [http://dx.doi.org/10.1016/S0002-9343(88)80234-1] [ Links ]
9. Ettinger WH, Klinefelter HF, Kwiterovitch PO. Effect of short-term, low-dose corticosteroids on plasma lipoprotein lipids. Atherosclerosis 1987;63(2-3):167-172. [http://dx.doi.org/10.1016/0021-9150(87)90117-1]
10. Ettinger WH Jr., Hazzard WR. Prednisone increases very low density lipoprotein and high density lipoprotein in healthy men. Metabolism 1988;37(11):1055-1058. [http://dx.doi.org/10.1016/0026-0495(88)90067-4]
11. Scherbakova IA, Gerasimova EN, Perova NV, Titova VN, Koldaeva AP, Galakhova IE. [Effect of hydrocortisone on lipid composition of blood serum lipoproteins in the development of experimental atherosclerosis]. Vopr Med Khim 1975;21(6):589-595. [ Links ]
12. Staels B, van Tol A, Chan L, Verhoeven G, Auwerx J. Variable effects of different corticosteroids on plasma lipids, apolipoproteins, and hepatic apolipoprotein mRNA levels in rats. Arterioscler Thromb 1991;11(3):760-769. [http://dx.doi.org/10.1161/01.ATV.11.3.760] [ Links ]
13. Reaven EP, Kolterman OG, Reaven GM. Ultrastructural and physiological evidence for corticosteroid-induced alterations in hepatic production of very low density lipoprotein particles. J Lipid Res 1974;15(1):74-83. [ Links ]
14. Bagdade JD, Yee E, Albers J, Pykalisto OJ. Glucocorticoids and triglyceride transport: Effects on triglyceride secretion rates, lipoprotein lipase, and plasma lipoproteins in the rat. Metabolism 1976;25(5):533-542. [http://dx.doi.org/10.1016/0026-0495(76)90007-X] [ Links ]
15. Mahley RW. Apolipoprotein E: Cholesterol transport protein with expanding role in cell biology. Science 1988;240(4852):622-630. [http://dx.doi.org/10.1126/science.3283935] [ Links ]
16. Lin RC. Effects of hormones on apolipoprotein secretion in cultured rat hepatocytes. Metabolism 1988;37(8):745-751. [http://dx.doi.org/10.1016/0026-0495(88)90009-1] [ Links ]
17. Hazra A, Pyszczynski NA, DuBois DC, Almon RR, Jusko WJ. Modeling of corticosteroid effects on hepatic low-density lipoprotein receptors and plasma lipid dynamics in rats. Pharm Res 2008;25(4):769-780. [http://dx.doi.org/10.1007/s11095-007-9371-8] [ Links ]
18. Nanjee MN, Miller NE. Plasma lipoproteins and adrenocortical hormones in men - positive association of low density lipoprotein cholesterol with plasma cortisol concentration. Clin Chim Acta 1989;180(2):113-120. [http://dx.doi.org/10.1016/0009-8981(89)90342-2] [ Links ]
19. Brotman DJ, Girod JP, Garcia MJ, et al. Effects of short-term glucocorticoids on cardiovascular biomarkers. J Clin Endocrinol Metab 2005;90(6):3202-3208. [http://dx.doi.org/10.1210/jc.2004-2379] [ Links ]
20. Atger V, Leclerc T, Cambillau M, et al. Elevated high density lipoprotein concentrations in heart transplant recipients are related to impaired plasma cholesteryl ester transfer and hepatic lipase activity. Atherosclerosis 1993;103(1):29-41. [http://dx.doi.org/10.1016/0021-9150(93)90037-U] [ Links ]
21. Choi HK, Seeger JD. Glucocorticoid use and serum lipid levels in US adults: The Third National Health and Nutrition Examination Survey. Arthritis Rheum 2005;53(4):528-535. [http://dx.doi.org/10.1002/art.21329] [ Links ]
 Correspondence:
Correspondence:
IL Ross
(ian.ross@uct.ac.za)
Accepted 30 April 2014


{kind=link}