










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSAMJ: South African Medical Journal
On-line version ISSN 2078-5135
Print version ISSN 0256-9574
SAMJ, S. Afr. med. j. vol.104 n.4 Pretoria Apr. 2014
CONTINUING MEDICAL EDUCATION
ARTICLES
N MankahlaI;A FigajiII
IMB ChB. Paediatric Neurosurgery Unit, Division of Neurosurgery, University of Cape Town and Red Cross War Memorial Children's Hospital, Cape Town, South Africa
IIMB ChB, PhD, FCNeurosurg (SA). Paediatric Neurosurgery Unit, Division of Neurosurgery, University of Cape Town and Red Cross War Memorial Children's Hospital, Cape Town, South Africa
ABSTRACT
Occult spinal dysraphism refers to a diverse group of congenital abnormalities resulting from varying degrees of disordered neuro-embryogenesis. Several terms have been used to describe these conditions, including spina bifida occulta and closed neural tube defects.
By definition, occult spinal dysraphism (OSD) is characterised by intact overlying skin, although most patients have some form of cutaneous stigmata. Because the underlying pathology is so variable, the clinical presentation and course of the condition vary significantly from severely disabling to asymptomatic throughout life. Management usually requires a multidisciplinary team of paediatricians, paediatric neurosurgeons, urologists, orthopaedic surgeons, occupational therapists, physiotherapists and geneticists. Although the spectrum of dysraphic abnormalities includes cranial anomalies (encephalocele), this article will focus on the spinal manifestations.
Epidemiology
Unlike open dysraphism, the exact prevalence of occult dysraphic disorders is unclear, in part because many patients may remain asymptomatic. The literature reports a combined incidence of spina bifida (open and closed) between 0.5 and 2.5/1 000 live births, varying among regions and populations. The incidence of OSD depends on how frequently relatively minor abnormalities are investigated and how often conditions are detected coincidentally. Pre-conception maternal folate consumption seems to have less of an effect than in open defects, although folate-dependent pathways in neural tube closure are involved in OSD. An association with maternal diabetes mellitus and anti-epileptic medications such as valproic acid and carbamazepine has been demonstrated. Genetic factors play significant predisposing roles, and much research has examined genes involved in folic acid metabolism pathways.[1] One such gene is the 5,10-methylenetetrahydrofolatereductase (MTHFR) gene located on chromosome 1. Folate is known to participate as a single-carbon donor for synthesis of thymidine and purines, the building blocks of DNA, and it is a methyl donor for methionine formation.[2,3] However, its exact role remains unclear.
Embryology
A clear understanding of the clinical and radiopathological spectrum of OSD requires an appreciation of the basic embryology of the nervous system. This will be discussed briefly below.
The development of the nervous system can be divided into three stages: gastrulation, primary neurulation and secondary neurulation.
Gastrulation
This begins by the end of the second week of gestation. The blastocyst, being a disc with two cell layers, is converted to a trilaminar plate by the formation of a third layer that will be the future mesoderm. This is triggered by the appearance of a primitive streak towards which cells migrate and which they pass through. It is also the first indication of a rostro-caudal orientation related to Hensen's node.
During this phase, there is a transient communication between the amnion and yolk sac, called the neurenteric canal.
Primary neurulation
At the end of the third week, the notochord initiates the differentiation of ectodermal cells into specialised neuro-ectoderm and the neural plate subsequently folds to form a tube. Closure of the neural tube occurs at up to five points in the human embryo.[4]
After closure, the neuro-ectoderm separates from the cutaneous ectoderm (dysjunction), and the space between is filled by mesoderm. This only accounts for the spine down to the S2 and S3 caudal levels. The remainder of the sacrococcygeal elements form subsequently.
Secondary neurulation
A caudal cell mass composed of pluripotent cells ascends from the tail bud of the embryo and, through a process of vacuolisation, forms a central canal that connects to the rostral cord formed during primary neurulation. The tip regresses, leaving in its wake the small filum terminale. Occult dysraphic abnormalities can be traced to a focal defect in one or more of these stages.[5]
Embryopathology
Table 1 lists the various pathologies that develop at different embryological stages. Open dysraphism largely results from primary neurulation, principally neural tube closure. In contrast, OSD involves any of the stages.
Spinal lipomas and dermal sinus tracks are the most common disorders presenting as clinical problems. Secondary neurulation lesions are the ones most frequently associated with anomalies of other germ-layer structures. Rarely, these pathologies are seen as part of a multiple malformation syndrome such as VACTERL (vertebral, anorectal, cardiac, tracheo-oesophageal fistula, oesophageal atresia, renal and limb defects).
The primary clinical problem in all these children is a tethered spinal cord, which is the key concept in understanding why they usually require treatment. The embryological problem occurs when the developing spinal cord lies low in the spinal canal. Normally, the conus of the spinal cord ascends in the spinal canal during fetal growth, as the vertebral column grows disproportionately in length. In OSD, however, the pathology (usually fat) attaches to the spinal cord low in the lumbosacral area, causing it to be tethered distally and progressively stretched (Fig. 3). Often there is a neurological deficit related to the failure of segmental development; however, sometimes the findings on neurological examination are normal. The primary concern, however, is that late neurological deterioration may develop (at any time from the first few months of life through to adulthood) as a consequence of mechanical stretch and ischaemic injury.
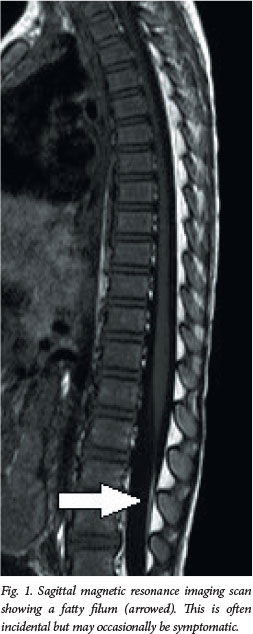
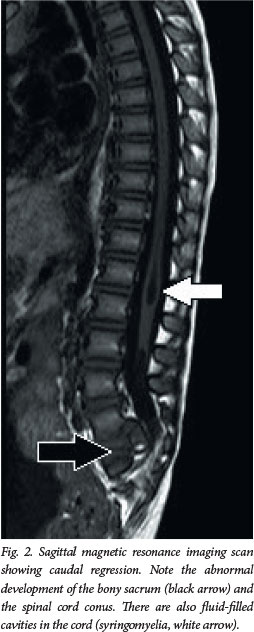
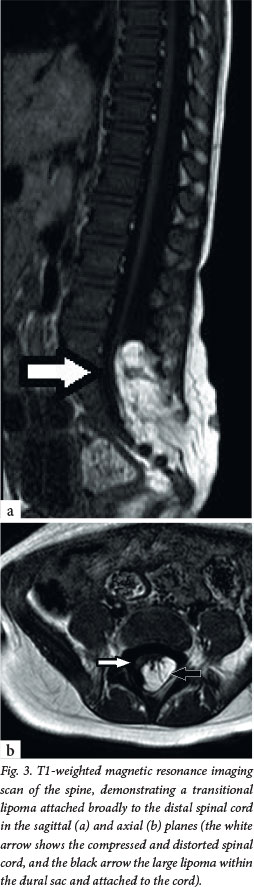
Clinical presentation
A detailed history should include a family history of associated abnormalities, maternal drug use during pregnancy, and pre-conception folate consumption. Recurrent urinary tract infections may signify a neurogenic bladder. Bed wetting or urge incontinence may be due to a spinal abnormality or simply a stage in development.
However, if these occur after the age of 5 years or are associated with other symptoms or signs, they should be investigated.
Previous meningitis (especially recurrent) may suggest a dermal sinus. Often the findings on examination of the newborn are apparently normal, but it must be remembered that neurological examination at this stage is relatively insensitive to subtle upper motor and lower motor neuron signs, both of which may reflect a static or progressive neurological deficit. Wasting of muscle groups is occasionally noticed, and the anus may demonstrate low external sphincter tone.
Cutaneous stigmata
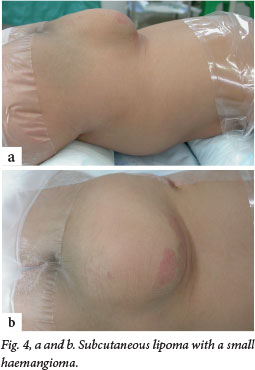
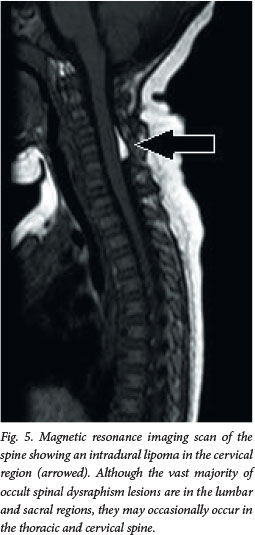
Although this disorder is termed occult, about 50 - 86% of patients will have cutaneous stigmata of dysraphism (Fig. 4). These are usually the presenting signs that initiate further investigation. The likelihood of positive imaging findings associated with cutaneous lesions increases significantly when a patient has two or more lesions.[6] The lesions are usually located in the midline lumbar region; occasionally they may be off-centre or further rostral along the spine (Fig. 5). Table 2 lists some of the abnormalities that may be noted, although any cutaneous lesion along the spine should raise concern about OSD.

Orthopaedic deformities
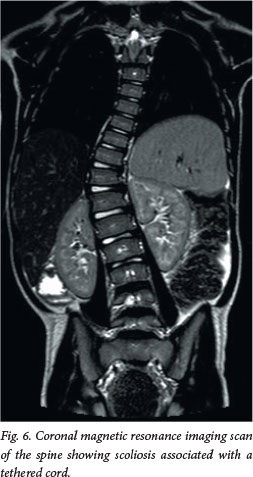
Approximately 50 - 70% of patients with OSD will have some form of orthopaedic deformity, including limb length discrepancy, unilateral atrophy, scoliosis (Fig. 6) and talipes equinovarus (club foot). In the toddler, a shortened, tight Achilles tendon may sometimes be the only sign. If a patient has an anorectal malformation, it is routine practice to perform a spinal magnetic resonance imaging (MRI) scan to exclude an associated tethered spinal cord.
Investigation and diagnostics
If OSD is suspected, the child should be referred for neurosurgical evaluation, preferably by a paediatric neurosurgeon, and the diagnosis must be made with MRI. Spinal ultrasound can be performed up to the age of 3 months, but it is unreliable as a stand-alone diagnostic tool because it is user-dependent and has a high false-negative rate. Spinal radiographs may reveal a defect of the spinous processes, scoliosis and hemivertebrae, but are seldom useful on their own. Ultrasound of the urogenital system is an absolute requirement to detect associated renal anomalies and signs of a neurogenic bladder.[8] Urodynamic testing should be performed on all patients with dysraphism to detect more subtle deficits.
MRI is the definitive imaging modality. It is non-invasive, non-irradiating and can image the whole spine. T1-weighted sagittal and axial images best demonstrate spinal lipomas and the fat-infiltrated thick filum terminale (fatty filum). Special highresolution sagittal sequences through the lesion assist in planning surgery. The brain is routinely imaged also - although findings are usually normal, occasionally imaging reveals a hindbrain abnormality. Even bony anatomy can be seen with sufficient detail to negate the need for computed tomography scanning.[9] We usually perform MRI between 3 and 6 months of age unless there is a reason to do it sooner. The quality of spinal MRI in the first few months of life is often suboptimal, and we generally operate at or after 6 months.
Management
Indications for surgery depend on the clinical presentation and the underlying pathology. The approach to tethering by an intradural lipoma depends on the presence of symptoms.
Management of the asymptomatic patient who presents with cutaneous stigmata alone is controversial. Some surgeons advocate a conservative approach, reserving surgery for those who develop symptoms. However, operating only when symptoms develop may not reverse the neurological deficit that has developed. Others advocate surgery if there is a complex tethering element, even if the patient is asymptomatic, as the likelihood of progression is high. However, this decision must be made carefully and in consultation with the parents because there is a risk of surgery-induced neurological injury. Regardless of the clinical scenario, we therefore recommend that all children who have surgery to untether the spinal cord (with the exception perhaps of a fatty filum) should have the operation performed under intra-operative neurophysiological monitoring control, and by a surgeon familiar with the condition. When surgery is indicated, we usually operate between the ages of 6 and 9 months unless there is progressive deterioration before this time.
For the symptomatic patient, the decision to operate is more straightforward.
In patients with split-cord malformations, the cord is usually tethered by an osteocartilagenous spur and surgical intervention is almost inevitable because symptoms progress over time.
Lipomyelomeningoceles and myelocystoceles, where the cord usually exits the spinal canal dorsally, almost always require surgery.
A thick and/or fatty filum, with or without a low-lying conus medullaris, is common in asymptomatic individuals. Often these are discovered incidentally. When the patient is symptomatic, sectioning is performed. If surgery is not indicated, bladder investigations and clinical follow-up are advisable.
Conclusion
Although OSD is less obvious than the open variant, it has a significant impact on function and therefore on the lives of both patient and family. A high index of suspicion is needed in order to avoid a missed diagnosis. If OSD is suspected, the patient should be referred to a paediatric neurosurgeon.
References
1. Bassuk AG, Kibar Z. Genetic basis of neural tube defects. Semin Pediatr Neurol 2009;16(3):101-110. [http://dx.doi.org/10.1016/j.spen.2009.06.001] [ Links ]
2. Botto L. Folic acid. Am J Epidemiol 2000;151(9):862-877. [http://dx.doi.org/10.1093/oxfordjournals.aje.a010290]
3. De Marco P, Merello E, Calevo MG, et al. Evaluation of a methylenetetrahydrofolate-dehydrogenase 1958G>A polymorphism for neural tube defect risk. J Hum Genet 2006;51(2):98-103. [http://dx.doi.org/10.1007/s10038-005-0329-6]
4. Kibar Z, Capra V, Gros P. Toward understanding the genetic basis of neural tube defects. Clin Genet 2007;71(4):295-310. [http://dx.doi.org/10.1111/j.1399-0004.2007.00793.x] [ Links ]
5. Thompson DNP. Spinal dysraphic anomalies: Classification, presentation and management. Paediatr Child Health (Oxford) 2010;20(9):397-403. [http://dx.doi.org/10.1016/j.paed.2010.03.011]
6. Schropp C, Sörensen N, Collmann H, Krauss J. Cutaneous lesions in occult spinal dysraphism -correlation with intraspinal findings. Childs Nerv Syst 2006;22(2):125-131. [http://dx.doi.org/10.1007/s00381-005-1150-4] [ Links ]
7. Drolet B. Cutaneous signs of neural tube malformations. Semin Cutan Med Surg 2004;23(2):125-137. [http://dx.doi.org/10.1016/j.sder.2004.01.006] [ Links ]
8. Lavallée LT, Leonard MP, Dubois C, Guerra LA. Urodynamic testing - is it a useful tool in the management of children with cutaneous stigmata of occult spinal dysraphism? J Urol 2013;189(2):678-683. [http://dx.doi.org/10.1016/j.juro.2012.08.203] [ Links ]
9. Tortori-Donati P, Rossi A, Cama A. Spinal dysraphism: A review of neuroradiological features with embryological correlations and proposal for a new classification. Neuroradiology 2000;42(7):471-491. [http://dx.doi.org/10.1007/s002340000325]
 Corresponding author:
Corresponding author:
N Mankahla
(ncefactor@ananzi.co.za)


{kind=link}