










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSAMJ: South African Medical Journal
On-line version ISSN 2078-5135
Print version ISSN 0256-9574
SAMJ, S. Afr. med. j. vol.103 n.12 Pretoria Dec. 2013
MOLECULAR GENETICS
Diagnostic, carrier and prenatal genetic testing for fragile X syndrome and other FMR-l-related disorders in Johannesburg, South Africa: A 20-year review
F B EssopI; A KrauseII
IMSc (Med). Division of Human Genetics, School of Pathology, Faculty of Health Sciences, University of the Witwatersrand, Johannesburg, South Africa, Division of Human Genetics, National Health Laboratory Service, Johannesburg, South Africa
IIMB BCh, PhD. Division of Human Genetics, School of Pathology, Faculty of Health Sciences, University of the Witwatersrand, Johannesburg, South Africa, Division of Human Genetics, National Health Laboratory Service, Johannesburg, South Africa
ABSTRACT
BACKGROUND: Fragile X syndrome (FXS), the most common inherited cause of intellectual disability (ID) worldwide, is caused by the expansion of a CGG repeat in the fragile X mental retardation gene (FMR-1) gene.
OBJECTIVES: To review, retrospectively, the genetic services for FXS and other FMR-1 -related disorders - including fragile X-associated tremor/ataxia syndrome (FXTAS) and FMR-1 -related primary ovarian insufficiency (POI) - at the Division of Human Genetics, Johannesburg, for diagnostic, carrier and prenatal genetic testing.
METHODS: The records of 2 690 patients who had genetic testing for FMR-1 between 1992 and 2012 were reviewed. Of these, 2 239 had diagnostic testing, 430 carrier or cascade testing and 17 prenatal testing for FXS. Four had FXTAS or POI testing. Polymerase chain reaction (PCR) and/or Southern blotting techniques were used to test the patients' samples for FMR-1 and FMR-2 expansions.
RESULTS: Of the 2 239 patients who had diagnostic FMR-1 testing, 128 (5.7%) had a full mutation, 12 (0.5%) had a premutation and 43 (1.9%) an intermediate allele. In 17 prenatal tests, eight fetuses tested positive for FXS. FMR-1 CGG repeat distribution analysis in 1 532 males negative for the FMR-1 expansion showed that 29 and 30 CGG repeats were the most common (61.1%), but the distribution was significantly different in the black and white populations.
CONCLUSION: The findings support the presence of FXS, as the most common cause of ID, in all local populations. The FMR-1 CGG repeat distribution varied from that found in other studies. The number of family members tested was relatively low suggesting that many at-risk individuals are not being referred.
Intellectual disability (ID) has an estimated prevalence of 2 - 3% in developed countries, but the prevalence of ID and its epidemiology in developing countries is not well established.[1] However, a study in a rural South African (SA) population reported the prevalence to be 3.6%.[2] The most common cause of ID in SA is fetal alcohol syndrome, which occurs at the highest rate in the world and at epidemic proportions in some communities in the Western Cape Province.[3] Fragile X syndrome (FXS) is the most common inherited cause of ID in all populations.
FXS is an X-linked neurodevelopmental disorder, with a variable phenotype in males and females, associated with intellectual, physical and behavioural features. ID is the most consistent feature, ranging from mild to severe, while the other clinical features include developmental delay, long narrow face, large prominent forehead and chin, protruding ears, macro-orchidism in post-pubertal males, hyperactivity, hand flapping, attention deficit and autism.[4] FXS is estimated to have a prevalence of approximately 1/4 000 in males and 1/8 000 in females worldwide.[5]
Almost all cases of FXS are caused by the expansion of a CGG repeat in the 5' untranslated region of the fragile X mental retardation gene (FMR-1) (OMIM #309550) at the FRAXA fragile site Xq27.3. A small number of deletions and missense mutations have also been reported. The CGG repeat size varies from 5 to 44 repeats in unaffected individuals. Intermediate (IM) alleles ('gray zone') range from 45 to 54 CGG repeats. The clinical significance of carrying an allele in this size range is unclear. Premutation (PM) carriers have expansions of 55 to 200 repeats, which are not associated with ID. Affected individuals have >200 CGG repeats and are classified as having a full mutation (FM) which is associated with hypermethylation of an upstream gene promoter region (CpG island) and silencing of FMR-1 gene transcription which results in the absence of the gene product, fragile X mental retardation protein (FMRP).[6]
FMR-1 -related disorders include FXS and two other clinically distinct conditions, fragile X-associated tremor/ataxia syndrome (FXTAS) (OMIM #300623) and FMR-1-related primary ovarian insufficiency (FXPOI) (OMIM #311360). Both males and females with a PM in FMR-1 have an increased risk of developing FXTAS, a progressive neurodegenerative disorder with a prevalence of 45% reported in males older than 50 years of age. Female PM carriers are less likely to develop FXTAS but have a 21% risk of developing primary ovarian insufficiency (POI) (menopause before the age of 40 years).'61
The FMR-1 gene, identified in 1991, is 38 kb long and consists of 17 exons which are alternatively spliced. It codes for the cytoplasmic protein FMRP, a631 amino acid RNA-binding protein that is expressed in almost every tissue, with highest levels observed in the brain and testes. FMRP has been shown to down-regulate the translation of specific target messenger RNAs and plays a vital role in synaptic plasticity.[7]
In 1981, the first SA cases of FXS were described in four families investigated by cytogenetic studies. Eleven affected males and one carrier female were found to have a fragile site on the X chromosome (fra(X)(q27)).[8] Cytogenetic detection of FRAXA is not reliable as a diagnostic test for FXS as it cannot differentiate between this and other fragile sites, such as FRAXD, FRAXE and FRAXF, and it is costly. Goldman et al.[9,10] presented the first molecular evidence that FXS occurs in the SA black ID population. They found nine (6.1%) FXS cases in 148 patients with ID, suggesting that the condition had been underdiagnosed in the past.
DNA testing for FXS has been available in SA since 1994 at the Division of Human Genetics (DHG), National Health Laboratory Service (NHLS) and the University of the Witwatersrand, Johannesburg. Prior to this, the diagnostic test was based exclusively on cytogenetic detection of the FRAXA fragile site. Currently, testing is performed by duplex polymerase chain reaction (PCR) analysis to size the normal and lower PM FMR-1 alleles, followed by Southern blotting to confirm expansions. The PCR assay is reliable, relatively cheap, and an efficient test as a primary screen for FXS. Mosaic males with a normal allele and an FM will yield a false-negative result.
Objective
To review and analyse the findings from DNA testing for diagnostic, carrier and prenatal purposes for FXS and other FMR-1-related disorders, conducted at the DHG over a 20-year period. The study provided a follow-up to a previous study by Goldman[11] in which FXS testing was evaluated by the DHG. The presence of FRAXE mutations and the distribution of FMR-1 and FMR-2 repeat sizes in the ID cohort negative for FXS were also determined as part of the present study.
Methods
Subjects
Patients tested for FXS and FMR-1-related disorders
A total of 2 690 individuals from 2 243 families underwent genetic testing for FXS and other FMR-1 -associated disorders through the DHG during the 20-year period (January 1992 - July 2012). Of these, 2 239 unrelated individuals (887 white, 959 black, 211 mixed ancestry and 182 Indian) had a suspected diagnosis of FXS or ID of unknown cause (1 961 males and 278 females), 430 were extended family members referred for FXS carrier or diagnostic testing (mothers of affected individuals, siblings and other at-risk second and third-degree relatives), four requested POI or FXTAS testing, and 17 were prenatal samples (obtained by chorionic villus sampling (CVS) or amniocentesis) from at-risk pregnancies.
The majority of referrals were from the Assessment and Learning Clinics at the Johannesburg (now the Charlotte Maxeke) Academic Hospital, Coronation (now Rahima Moosa) Hospital, Alexandra Clinic and institutions for people with ID. Further referrals were from local general practitioners, paediatricians and medical geneticists at the Genetic Counselling Clinics (at Chris Hani Baragwanath Academic Hospital, Johannesburg Hospital, Donald Gordon Medical Centre and Coronation Hospital) and genetic nurses. However, a small number of cases were referred from other genetic centres around SA and from doctors in private practice. Blood samples from the patients were sent in with a referral form stating the possible diagnosis and, in some cases, the clinical features observed. Information collected from patient files included (where available) each subject's gender, ethnicity, their test results and pregnancy information (where applicable).
DNA extraction
Genomic DNA was extracted from whole blood using a salting out method or a commercial DNA extraction kit (High Pure PCR Template Preparation Kit, Roche Diagnostics) for blood samples <1 ml. DNA was extracted from CVS and amniocyte material using a phenol-chloroform extraction method.
PCR analysis
PCR amplification was performed using published primers'12,131 with detection of products on the ABI PRISM 377 or ABI 3130xl Genetic Analyzer (Applied Biosystems). Testing for AGG interruptions was not performed.
Southern blot analysis
Southern blot analysis was performed to establish the methylation status of the promoter region, to detect FMs that failed to amplify on PCR analysis and to differentiate females who were homozygous for a normal-sized FMR-1 allele from female PM/FM heterozygotes. Analysis of the FMR-1 expansion was carried out by digestion of genomic DNA (5 - 15 µg) with both EcoRI (Roche) and a methylation-sensitive enzyme, EclXI (Roche). Hybridisation was performed using the StB12.3 probe and labelled with the radionuclide isotope a-32PdCTP using the Megaprime DNA Labelling Systems kit (Amersham).
Results
FMR-1 expansion
Total cohort
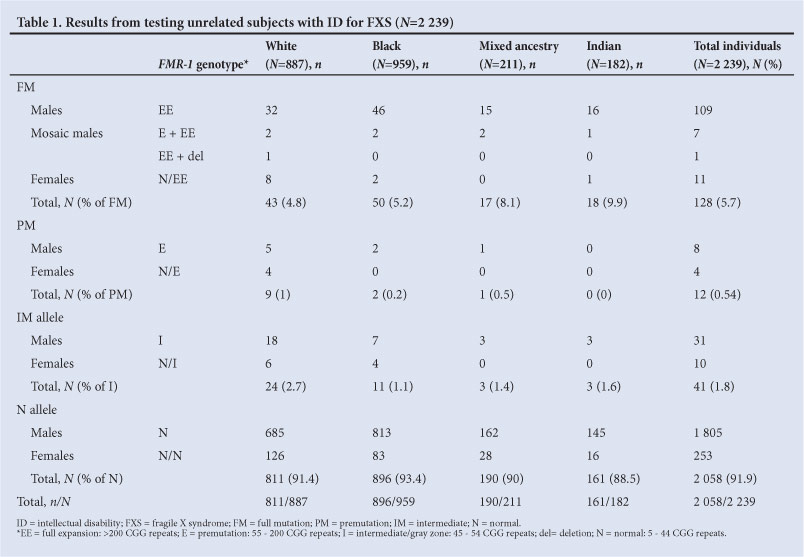
Of the 2 239 unrelated individuals with ID, 128 (5.7%) tested positive for the FMR-1 FM (117; 5.2% male and 11; 0.5% female probands). Of these, the FM was observed in 4.8% (43/887) of white patients, 5.2% (50/959) of black, 8.1% (17/211) of mixed ancestry and 9.9% (18/182) of Indian patients. Chi-squared analysis showed no significant difference (p=0.72) between the observed frequencies of FMs in the black and white populations (Table 1).
PMs accounted for 0.54% (12/2 239) of the total diagnostic cohort, ranging from 0% to 1% in the different ethnic groups tested; while IM alleles were observed in 1.8% (41/2 239) of the cohort, ranging from 1.1% to 2.7% in the different ethnic groups tested.
Cascade testing
Of the 430 extended family members tested in the 2 243 families, an FM was found in 51 (11.8%; 22 males; 29 females) subjects, a PM and/or IM in 70 (16.3%; 59 females; 11 males) and 309 (71.9%) individuals tested negative.
FXTAS and POI referrals
A PM was found in 1/3 females tested, and one male referred for FXTAS tested negative.
Prenatal diagnostic testing
A total of 17 prenatal tests (12 white, four Indian, one black and none of mixed ancestry) were performed on CVS material or amniotic cells. Eight of the 17 (47%) fetuses had an FMR-1 FM (five male and three female), two a PM (one male and one female) and seven (41%) tested negative.
Incidental findings
Female siblings with FMR-1 expansions of both alleles
A large family positive for the FMR-1 expansion (n=24) was investigated and two female siblings were identified as having an FMR-1 expansion on both X chromosomes. Both siblings were found to have one IM allele of 51 CGG repeats and a second PM allele of 57 repeats in one sibling and 69 repeats in the other. Both siblings inherited the 51 repeat unchanged from their father while the 57 repeat from their mother was inherited stably in one, but was shown to have expanded to 69 repeats in the other, and subsequently it expanded to an FM in her grandson.
FRAXE mental retardation syndrome
Amplification of the FMR-2 GCC repeat was used as an internal control primer in the PCR assay. An expansion of >200 GCC repeats in the FMR-2 gene at Xq28 has been shown to cause FRAXE syndrome (OMIM #309548), a rare condition associated with a mild form of ID.[14] Of the 1 961 males with ID tested on the duplex PCR assay, FMR-2 results were obtained for 1 558 subjects (who tested negative for the FMR-1 expansion). None were found to have an FMR-2 FM. However, an IM allele (26 - 59 GCC repeats) was observed in four males (0.3%).
Distribution of FMR-1 CGG repeat sizes
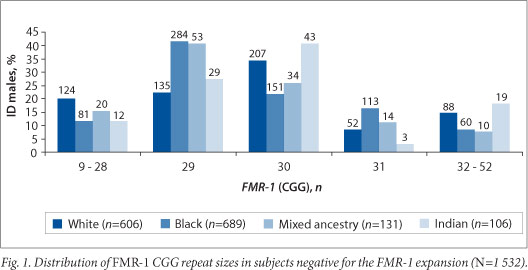
The distribution of the CGG repeat size in 1 532 FMR-1 expansion-negative ID subjects is shown in Fig. 1. The smallest FMR-1 CGG repeat size detected was 9. The most frequent repeat size observed was 29 CGG repeats (32.7%; 501/1 532) followed by 30 CGG repeats (28.4%; 435/1 532). CGG repeat sizes 20, 23, 24, 29, 30, 31 and 32 together accounted for the majority (86%; 1 317/1 532) of alleles in the study population. The most common allele observed in the black and mixed-ancestry population was 29 CGG repeats (the frequency was 41%; 284/689 and 53/131, respectively), while 30 CGG repeats was the most common number in the white (34%; 207/606) and Indian populations (41%; 43/106). Chi-squared analysis showed a significant difference (p<105) in the frequency of the 29 and 30 CGG repeat sizes in the black and white populations.
Distribution of FMR-2 GCC repeat sizes
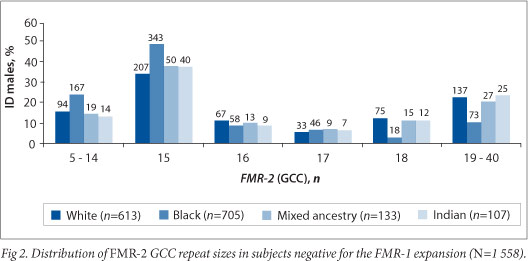
The distribution of the GCC repeat size in 1 558 FMR-1 -expansion-negative subjects is shown in Fig. 2. FMR-2 allele sizes ranged from 5 to 40 GCC repeats, with 15 being the most common repeat size, accounting for 41% (640/1 558) of alleles identified. The distribution was evident in each of the four ethnic groups, and observed at a frequency of 33.8% (207/613) in whites, 48.7% (343/704) in blacks, 37.6% (50/133) in mixed ancestry and 37% (40/108) in Indians. Chi-squared analysis showed a significant difference (p<105) between the distributions of FMR-2 alleles in the black and white populations.
Discussion
The present study reviewed the molecular genetic testing for FXS and other FMR-1 -related disorders conducted over a 20-year period at the DHG, NHLS, Johannesburg.
In 2000, the PCR assay was introduced as an initial screen for the routine diagnosis of FXS. This has improved and refined the diagnostic testing by accurate determination of the FMR-1 CGG repeat size, which was not possible with Southern blotting. It has also reduced the number of Southern blots required and decreased the turnaround time for patients with CGG repeat sizes up to 120 repeats.
Full mutations in the total cohort
An FMR-1 FM was identified in 5.7% (128/2 239) of the probands tested, comparable with findings from ID cohorts worldwide which show FXS estimates ranging from 0.5% to 5%, depending on how cohorts were determined.[15] Our study provides further evidence to support that of Goldman et al.[9] that FXS, previously thought to be rare in the SA black population, is underdiagnosed.
FMs were observed in 5.2% (50/959) of all black subjects tested, which is slightly lower than that found in Goldman et al's small sample (nine affected in 148 patients; 6.1%), but may be considered to be a more accurate reflection due to the larger sample involved. This result lends further support to the findings of Goldman et al.[9] that FXS is a common cause of ID in all SA ethnic groups.
In the Indian population, FMs were found in 9.9% (18/182) of referrals for ID, which is higher than that observed in the other three ethnic groups. The majority of these Indian probands were referred from KwaZulu-Natal, and a selection bias is possible, as testing is expensive and referrals may be limited to patients with a high clinical suspicion of FXS. Interestingly, studies in the Indian population on the Indian subcontinent report a frequency of approximately 7.48% for FXS FMs,[16] which is lower than the frequency observed in the present study but higher than worldwide figures. The frequency of FMs in the mixed-ancestry population was 8.1%, which is also higher than the frequency observed in the white and black ethnic groups, but the sample size was small and the differences were not statistically significant.
Premutations and intermediate alleles in the ID cohort
In the total cohort, a PM was detected in 0.54% (12/2 239) while an IM allele was detected in 1.83% (41/2 239). Both PMs and IM alleles were observed at higher frequencies within the white ID population (1% and 2.7%, respectively) compared with the other populations. PMs are estimated to occur in the general population at a prevalence of ~1/113 - 1/259 among females (~0.5%) and ~1/260 - 1/800 (~0.2%) among males; i.e. much more common than FXS (1/4 000 males).[4] Further, a significant excess of IM alleles in boys with special education needs compared with normal controls has been reported. There is controversy around whether or not males and females with a PM have subtle behavioural or learning difficulties. However, a number of studies have suggested that there is no clinical significance to the presence of PMs or IMs in individuals with ID.[17]
Cascade screening of extended family members detected an additional 121 subjects with an FMR-1 FM, including mothers of affected individuals, siblings and other at-risk second- and third-degree relatives. As the FMR-1 mutation is rarely a new mutation, cascade screening from a proband is very worthwhile as it often detects other affected individuals in a family. The number of family members tested by cascade screening in the present study was low, suggesting that many at-risk family members are not being tested and/or offered appropriate genetic counselling.
Female siblings with FMR-1 expansions of both alleles
The family in which two females were found to have expansions of both alleles is the first such case described in SA and the fifth such family in the world.[18] Clinical data were not available on these females, but the two sisters would not be expected to present with ID. Mostly, studies have shown that females with two PMs do not have ID nor physical features typical of FXS, while those with one FM have characteristics typical of females with FXS.[18] They would be predicted to be at risk of POI and FXTAS, but the exact risk is unclear.
FRAXE syndrome
The present study confirms the rarity of FRAXE syndrome in SA, consistent with reports from other studies.[19] FRAXE syndrome has an estimated population prevalence of 1/23 500, based on two large population-based studies undertaken on special education needs individuals. A study on a cohort of white and African American subjects showed no FMR-2 expansion, while another report found one FRAXE syndrome-positive subject among 3 731 boys with ID.[19-FRAXE screening is therefore not indicated and testing has not been implemented routinely in the DHG. Amplification of the FRAXE allele is, however, useful in the duplex PCR FRAX assay as a control for PCR failure, especially in males, and as an indicator of an FMR-1 expansion. Its value has also been demonstrated as a linked marker in tracking the FMR-1 expansion in families, especially in prenatal testing, as it may reduce the time taken to obtain an initial result.
Distribution of FMR-1 alleles
Our study investigated the distribution of the CGG repeat in 1 532 FMR-1-expansion-negative ID subjects. The CGG repeat sizes of 29 and 30 were found to be most common in all ethnic groups, together accounting for 61.1% of the alleles in the cohort and comparable with populations of western European ancestry and other populations in Asia, Indonesia, Brazil, Chile, Cameroon, Ghana and India, and in African Americans. [20] In the present study the 30 CGG repeat (found in 34% of cases) was the most common allele in the white population, which corroborates well with the 39% frequency reported by Goldman.[11] The most common allele in the black population in this study was shown to be 29 CGG repeats (in 41% of the total black cohort). This finding differs from that of Goldman,[11] who showed that 28 CGG repeats (32%) was a common finding in SA black individuals. Our study is more likely to reflect the correct size as newer techniques have allowed for more accurate sizing. However, a study in a Cameroonian population also showed the 29 CGG repeat to be most common, whereas studies among African Americans and Ghanaians suggested that the 30 CGG repeat was the most common allele. Further studies are needed to distinguish true distribution differences from technical ones. Very few studies have been performed on African populations and the present study adds significantly to the African population data.
Distribution of FMR-2 alleles
This is the first study conducted on SA males with ID which provides information on the range of the FMR-2 GCC repeat sizes. FMR-2 allele distribution in most ID populations showed that the 15 GCC repeat was the most frequent allele, similar to that observed in our study. Four males in this sample were found to have an FMR-2 repeat size in the IM range. Studies have disputed the association of IMs with ID and suggested that more studies with larger numbers are needed to resolve this matter.[19]
Conclusion
This study strongly supports the fact that FXS is the most common cause of ID in all the local populations. However, FRAXE syndrome was not shown to contribute significantly to ID in the SA population and has therefore not been incorporated into the routine diagnostic testing. Earlier studies undertaken in our laboratory on X-linked mental retardation genes also suggested that mutations in ARX, XNP and HOPA like most others, are rare, and not significant contributors to ID locally.[21,22]
The presence of an FMR-1 expansion in a family member has significant implications, as there are likely to be other affected and at-risk relatives. A confirmed diagnosis leads to improved support for newly-diagnosed patients and potentially allows identification of all the carriers in the family. Prenatal and pre-implantation genetic diagnosis can be offered to at-risk individuals and this service allows couples to make informed choices about their future. Attempts should be made to expand offers of cascade testing once a proband is diagnosed. Comprehensive genetic counselling services need to become more widely available so that they can assist affected families with these issues.
Acknowledgements
The authors thank colleagues at the DHG Molecular Diagnostic Laboratory, who have performed the routine testing over the years. Funding for this study was provided from the NHLS Research Trust, the SA Medical Research Council and the HE Griffin Charitable Trust, University of the Witwatersrand.
References
1. Roeleveld N, Zielhuis GA, Gabreéls F. The prevalence of mental retardation: A critical review of recent literature. Dev Med Child Neurol 1997;39(2):125-132. [http://dx.doi.org/10.1111/j.1469-8749.1997.tb07395.x] [ Links ]
2. Kromberg J, Zwane E, Manga P, Venter A, Rosen E, Christianson A. Intellectual disability in the context of a South African population. JPPID 2008;5(2):89-95. [http://dx.doi.org/10.1111/j.1741-1130.2008.00153.x] [ Links ]
3. May PA, Gossage JP, Marais A-S, et al. The epidemiology of fetal alcohol syndrome and partial FAS in a South African community. Drug Alcohol Depend 2007;88(2-3):259-271. [http://dx.doi.org/10.1016/j.drugalcdep.2006.11.007] [ Links ]
4. Pirozzi F, Tabolacci E, Neri G. The FRAXopathies: Definition, overview, and update. Am J Med Genet 2011;155A(8):1803-1816. [http://dx.doi.org/10.1002/ajmg.a.34113] [ Links ]
5. Coffee B, Keith K, Albizua I, et al. Incidence of fragile X syndrome by newborn screening for methylated FMR1 DNA. Am J Hum Genet 2009;85(4):503-514. [http://dx.doi.org/10.1016/j.ajhg.2009.09.007] [ Links ]
6. Saul R, Tarleton J. FMR-1 related disorders. In: Pagon RA, Adam MP, Bird TD, et al, eds. GeneReviews. Seattle: University of Washington, 1993-2013. http://www.ncbi.nlm.nih.gov/books/NBK1384/ (accessed 9 May 2013). [ Links ]
7. Santoro MR, Bray SM, Warren ST. Molecular mechanisms of fragile X syndrome: A twenty-year perspective. Annu Rev Pathol 2012;7(1):219-245. [http://dx.doi.org/10.1146/annurev-pathol-011811-132457] [ Links ]
8. Venter PA, Gericke GS, Dawson B, Op't Hof J. A marker X chromosome associated with nonspecific male mental retardation. The first South African cases. S Afr Med J 1981;60(21):807-811. [ Links ]
9. Goldman A, Krause A, Jenkins T. Fragile X syndrome occurs in the South African black population. S Afr Med J 1997;87(4):418-420. [ Links ]
10. Goldman A, Jenkins T, Krause A. Molecular evidence that fragile X syndrome occurs in the South African black population. J Med Genet 1998;35(10):878. [http://dx.doi.org/10.1136/jmg.35.10.878] [ Links ]
11. Goldman, A. Trinucleotide Repeat Disorders in Southern African Populations: A Molecular Study. PhD thesis. Johannesburg: University of the Witwatersrand, 1997. [ Links ]
12. Fu YH, Kuhl DP, Pizzuti A, et al. Variation of the CGG repeat at the fragile X site results in genetic instability: Resolution of the Sherman paradox. Cell 1991;67(6):1047-1058. [http://dx.doi.org/10.1016/0092-8674(91)90283-5] [ Links ]
13. Wang Q, Green E, Bobrow M, Mathew CG. A rapid, non-radioactive screening test for fragile X mutations at the FRAXA and FRAXE loci. J Med Genet 1995;32(3):170-173. [http://dx.doi.org/10.1136/jmg.32.3.170] [ Links ]
14. Gecz J. The FMR2 gene, FRAXE and non-specific X-linked mental retardation: Clinical and molecular aspects. Ann Hum Genet 2000;64(2):95-106. [http://dx.doi.org/10.1046/j.1469-1809.2000.6420095.x] [ Links ]
15. Biancalana V, Beldjord C, Taillandier A, et al. Five years of molecular diagnosis of fragile X syndrome (1997-2001): A collaborative study reporting 95% of the activity in France. Am J Med Genet 2004;129A(3):218-224. [http://dx.doi.org/10.1002/ajmg.a.30237] [ Links ]
16. Kabra M, Sharma D, Singh D, et al. Fragile X screening for FRAXA and FRAXE mutations using PCR based studies: Results of a five year study. Ind J Hum Genet 2006;12(1):17. [http://dx.doi.org/10.4103/0971-6866.25297] [ Links ]
17. Sherman SL, Marsteller F, Abramowitz AJ, Scott E, Leslie M, Bregman J. Cognitive and behavioral performance among FMR1 high-repeat allele carriers surveyed from special education classes. Am J Med Genet 2002;114(4):458-465. [http://dx.doi.org/10.1002/ajmg.10303] [ Links ]
18. Linden MG, Tassone F, Gane LW, Hills JL, Hagerman RJ, Taylor AK. Compound heterozygous female with fragile X syndrome. Am J Med Genet 1999;83(4):318-321. [http://dx.doi.org/10.1002/(SICI)1096- 8628(19990402)83:4<318::AID-AJMG16>3.3.CO;2-P] [ Links ]
19. Crawford DC, Meadows KL, Newman JL, et al. Prevalence and phenotype consequence of FRAXA and FRAXE alleles in a large, ethnically diverse, special education-needs population. Am J Hum Genet 1999;64(2):495-507. [http://dx.doi.org/10.1086/302260] [ Links ]
20. Peprah E. Fragile X syndrome: The FMR1 CGG repeat distribution among world populations. Ann Hum Genet 2012;76(2):178-191. [http://dx.doi.org/10.1111/j.1469-1809.2011.00694.x] [ Links ]
21. Essop FB. Molecular Aspects of X-Linked Mental Retardation Loci. MSc (Med) dissertation. Johannesburg: University of the Witwatersrand, 2010. [ Links ]
22. Friez MJ, Essop FB, Krause A, et al. Evidence that a dodecamer duplication in the gene HOPA in Xq13 is not associated with mental retardation. Hum Genet 2000;106(1):36-39. http://dx.doi.org/10.1007/s004390051006] [ Links ]
 Correspondence:
Correspondence:
F B Essop
(fahmida.essop@nhls.ac.za)
Accepted 12 August 2013.


{kind=link}