










Serviços Personalizados
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkSAMJ: South African Medical Journal
versão On-line ISSN 2078-5135
versão impressa ISSN 0256-9574
SAMJ, S. Afr. med. j. vol.103 no.12 Pretoria Dez. 2013
MOLECULAR GENETICS
Biology and genetics of oculocutaneous albinism and vitiligo - common pigmentation disorders in southern Africa
P MangaI; R KerrII; M RamsayIII; J G R KrombergIV
IPhD. The Ronald O Perelman Department of Dermatology and the Department of Cell Biology, New York University School of Medicine, New York, USA
IIPhD. Division of Human Genetics, University of the Witwatersrand and National Health Laboratory Service, Johannesburg, South Africa
IIIPhD. Division of Human Genetics, University of the Witwatersrand and National Health Laboratory Service, Johannesburg, South Africa
IVBA (Soc Work), MA, PhD. Division of Human Genetics, University of the Witwatersrand and National Health Laboratory Service, Johannesburg, South Africa
ABSTRACT
Pigmentation disorders span the genetic spectrum from single-gene autosomal recessive disorders such as oculocutaneous albinism (OCA), the autosomal dominant disorder piebaldism to X-linked ocular albinism and multifactorial vitiligo. OCA connotes a group of disorders that result in hypopigmented skin due to decreased melanin production in melanocytes and loss of visual acuity. There are four non-syndromic forms, OCA1-4, which are classified based on the gene that is mutated (tyrosinase, OCA2, tyrosinase-related protein 1 and SLC45A2, respectively). Despite the fact that multiple genes account for the various forms of OCA, the phenotypes of all four forms result from disruption in the maturation and trafficking of the enzyme tyrosinase. OCA2 is the most prevalent autosomal recessive disorder among southern African blacks, affecting 1/3 900 individuals; while OCA3, although rare, is most prevalent in southern Africa. Another common pigmentation disorder in southern Africa is vitiligo, which affects 1 - 2% of people worldwide. Vitiligo is a complex, acquired disorder in which melanocytes are destroyed due to an autoimmune response. The aetiology underlying this disorder is poorly understood, although recent genetic association studies have begun to shed light on the contributing factors. Pigmentation disorders have significant psychosocial implications and co-morbidities, yet therapies are still lacking. Recent progress in our understanding of the pathobiology of both albinism and vitiligo may herald novel treatment strategies for these disorders.
Prashiela Manga joined the Department of Human Genetics at the University of Witwatersrand in 1991 as an Honours student and went on to complete a PhD in 1997. Both her projects focused on mapping genes involved in albinism. Her fascination with the biology of pigmentation was fostered by her mentors and co-authors on this review, MR and JK, and in no small part by interactions and discussions with Trefor Jenkins. PM's laboratory at the New York University School of Medicine continues to focus on unravelling the mechanisms involved in regulation of skin pigmentation and elucidating the pathobiology of various pigmentation disorders.
Jennifer Kromberg worked with Trefor Jenkins in his Department of Human Genetics from 1971 to 1999 and again later in the same department, as a retiree, from 2005 to date. As a social scientist she undertook both MA and PhD studies on the psychosocial and genetic aspects of oculocutaneous albinism, and has published many papers on the topic. During the early 1990s the albinism work required a molecular approach and this work was primarily guided by Michele Ramsay who had recently returned following a postdoctoral fellowship in London. Four molecular PhD projects on albinism were completed by students in the department: Mary-Anne Kedda (nee Colman), Gwyneth Stevens, Prashiela Manga and Robyn Kerr. All four have proceeded with careers in science and Robyn Kerr is currently an academic member of the Division. It is a great pleasure to write this review for the Festschrift in honour of Trefor Jenkins' remarkable academic career.
Biology of pigmentation
The primary pigment that determines human skin, hair and eye colour is melanin, which is synthesised by melanocytes. Melanin protects the skin from ultraviolet (UV) radiation and there is an inverse correlation between the degree of constitutive pigmentation and the risk of sun-induced skin cancers. Besides the life-threatening cancer risk, loss of pigmentation results in premature aging, compromised cutaneous immunity and significant emotional distress to affected individuals.[1-3]
During embryonic development, melanocyte precursors that arise from the neural crest populate several areas including the interfollicular epidermis and hair follicle bulge in the skin; the uveal tract of the eye; and the stria vascularis, vestibular apparatus and endolymphatic sac of the ear. The development of melanocytes is tightly regulated at the genetic level by a number of genes that control proliferation, survival and migration of precursor cells to the various sites of the body and their differentiation into active melanocytes. A key regulator of this process is the microphthalmia transcription factor (MITF), which has been dubbed the 'master regulator' of the melanocyte, capable of modulating expression of several melanocyte-specific proteins.[4]MITF mutations result in Waardenburg syndrome type II.[5] Once the melanocyte has differentiated, MITF regulates expression of genes in response to UV exposure, facilitating the tanning response.
Melanocytes produce two forms of melanin, black-brown eumelanin and red-yellow pheomelanin. Skin and hair colour is determined by the total amount and ratio of eumelanin to pheomelanin. Melanogenesis occurs within membrane-bound organelles known as melanosomes. This limits the potential for cellular damage by the intermediates of melanin synthesis, which include reactive oxygen species. Melanosomes consist primarily of proteins that are synthesised in the endoplasmic reticulum (ER) and are then routed to the melanosome either directly or via the Golgi where additional modifications, such as glycosylation, are made to the protein.
Mutations in a host of genes have been described that result in the failure of protein delivery to the melanosome. The majority of these comprise the various forms of Hermansky-Pudlak syndrome. To date, at least nine forms have been described, each due to mutations in a different gene. In addition to oculocutaneous albinism (OCA), affected individuals lack platelet-dense bodies, causing a bleeding diathesis, and can experience granulomatous colitis or fatal pulmonary fibrosis.[6]
Once the key enzymes, including tyrosinase, and structural proteins have been delivered, melanin synthesis begins. Melanin fills the melanosome, which is transported to the dendrites of the melanocyte and is eventually transferred to the keratinocytes where it forms a nuclear cap that protects DNA from UV-induced damage. In white skin, melanosomes are small, not very heavily pigmented and form membrane-bound clusters in the keratinocytes that are eventually degraded generating 'melanin dust' in suprabasal layers of the skin.[7] In black skin, melanosomes are large and remain as single organelles throughout the skin, while both forms are found in Asian skin.[8]
Over a hundred genes are thought to play a role in melanin synthesis and melanocyte function; however, only 11 have been confirmed as key determinants of normal skin pigment variation through genome-wide association studies to date,[9] with some genes linked exclusively to either inter- or intra-population variation. Fewer genes have been implicated in determining hair and eye colour, with the melanocortin-1 receptor (MC1R) shown to be the 'red hair' gene[10] and OCA2 the 'brown eyes' gene.[11]
OCA
OCA denotes a group of common autosomal recessive disorders resulting from disruption of melanin synthesis. There are four major forms: (i) OCA1 (mutated tyrosinase (TYR) gene);[12](ii) OCA2(mutated OCA2 gene);[13](iii) OCA3 (mutated tyrosinase-related protein 1 (TYRP1) gene);[14] and (iv) OCA4 (mutated solute carrier family 45, member 2 (SLC45A2) gene).[15] OCAs are characterised by decreased or absent melanin in skin, hair and eyes. Lacking photoprotection provided by melanin, individuals with OCA are highly susceptible to skin cancers, particularly squamous cell carcinoma.
A loss of visual acuity is also a consequence of OCA. During development, decreased melanin synthesis in the retinal pigment epithelium results in foveal hypoplasia and dysregulation of adjacent retinal ganglion cells, and consequently, misrouted decussation of the nerve fibres connecting the retina to the brain at the optic chiasm. Melanin also plays a role in the adult eyes where it is important for reducing light scatter and improving acuity. Individuals with OCA thus experience nystagmus, strabismus and photophobia. A significant number of children in southern African schools for the blind suffer from OCA.
In addition to the physiological issues, the highly visible nature of the OCA phenotype, particularly in black communities, causes significant psychosocial co-morbidities. These include maternal rejection in infancy,[16] later adjustment problems and stigmatisation due in part to the widely believed death myth that people with albinism do not die normal deaths, but disappear at the end of their lives.[17] A study of Nigerians with OCA2 found that they experienced alienation, avoided social interactions and were less emotionally stable. Furthermore, affected individuals were less likely to complete schooling, find employment and find partners.[18]
Albinism occurs at high frequencies in populations of African origin. OCA2 is particularly common, with a few mutations accounting for most cases, suggesting a shared genetic history.[20] Several factors may have contributed to the retention of albinism-causing mutations: (i) carriers with lighter skin are considered more suitable mates; (ii) fertility advantage; and (iii) carrier advantage such as reduced susceptibility to disease (e.g. lighter skin may result in fewer mosquito bites and a reduced risk of malaria).
Molecular and biochemical basis
The biochemical basis of albinism was first speculated by Garrod in 1908 as an inborn error of metabolism.[19] He correctly theorised that the phenotype was due to the absence of enzyme activity. The enzyme in question is tyrosinase, the key melanogenic enzyme that catalyses the first biosynthesis reaction converting tyrosine to L-3,4-dihydroxyphenylalanine (L-DOPA). The maturation process required to generate a functional tyrosinase enzyme is complex and highly regulated. Maturation begins in the ER where chaperones facilitate folding, post-translational modification and acquisition of tertiary structure. Further modifications are made in the Golgi before tyrosinase is transported to melanosomes. Mutations that result in OCA1-4 all result in retention of tyrosinase in the ER and/or misrouting that prevents delivery to the melanosome. One approach to promote tyrosinase maturation in melanocytes in culture is to increase the levels of tyrosine or L-DOPA, which accounts for an early observation, prior to genetic classification of albinism, that hairbulbs incubated in high levels of tyrosine could indicate whether the type of OCA was tyrosinase-negative (no increase in melanin) or -positive (heavily pigmented hairbulbs post incubation).
OCA1
OCA1 is an autosomal recessive disorder caused by TYR mutations. To date, more than 100 mutations have been described at this locus. OCA1 has been reported throughout the world and occurs with a frequency of about 1/40 000 worldwide, with the highest prevalence in white populations.[12] It is extremely rare in black populations.[21] OCA1 presents at birth with a range in severities. The most severe phenotype, OCA1A or tyrosinase-negative albinism, results from a complete lack of enzyme activity and pigment remains completely absent in the skin, hair and eyes throughout life. Hypopigmentation is accompanied by a severe ocular phenotype. Milder mutations encode proteins with some residual enzyme activity that result in slightly less severe phenotypes. These include OCA1B (affected individuals are born with white skin and hair but develop some pigment with age and express less severe ocular findings than in OCA1A), OCA1TS (tyrosinase is temperature-sensitive and active in cooler regions of the body resulting in a phenotype similar to that of the Siamese cat) and platinum OCA (small amounts of pigment accumulate in the hair and eyes in late childhood resulting in a silver tinge).[22] A recent study identified a potential therapeutic for OCA1 where some tyrosinase activity is present. Onojafe et al.,[23] having noted that nitisinone (used in the treatment of hereditary tyrosinemia type 1) caused an increase in serum tyrosine levels, treated OCA1B mice with the drug and noted an improvement in pigmentation of the mice.[23] This is the first potential therapeutic treatment of OCA and may even be of use in prenatal treatment to prevent the optic tract misrouting that results in loss of visual acuity. The effects of nitisinone on a developing fetus are not known; however, at least one patient has been reported to have had a successful pregnancy while taking nitisinone,[24] although no clinical studies have been performed to date.
OCA2
OCA2 is the most common form of albinism worldwide due to its high prevalence in southern Africa, where it occurs in 1/3 900 blacks.[21] Affected individuals are born with some pigmentation and there is a slight increase in pigmentation with age. Hair colour ranges from yellow to light brown, while the skin is white. In some families, affected individuals develop multiple freckles in sun-exposed areas. Ocular findings are generally less severe than those in OCA1A.
OCA2 results from mutations in the human homologue of the mouse pink-eyed dilution gene, OCA2 (formally known as the P gene). The OCA2 gene is predicted to encode a transport or channel protein,[25] although its precise function is not known. OCA2 was mapped to human chromosome 15q11-12 and showed evidence of locus homogeneity in black South African (SA) families with OCA2.[26] OCA2-associated haplotypes suggested multiple mutations and mutation analysis revealed one common mutation, a 2.7 kb deletion of exon 7, among these families.[20]
The origin of the deletion has been traced back to a founding mutation in central Africa.[20] In parts of Africa, including SA, about 80% of OCA2 chromosomes will carry the deletion, making it a useful diagnostic tool. A subsequent study investigating non-2.7 kb alleles in affected individuals from different regions in Africa (including SA, Lesotho, Zimbabwe and the Central African Republic) found no second common mutation.[27]
OCA2B, known as brown albinism (Fig. 1), is much milder than OCA1 and OCA2. It was first described in Nigeria,[28] but we have since shown that it is a milder form of OCA2 resulting from mutations in the OCA2 gene.[29] Individuals with brown albinism have a cream to tan skin, beige to light brown hair and blue-green to brown irides. Freckles, similar to those seen in OCA2, may develop in sun-exposed areas.
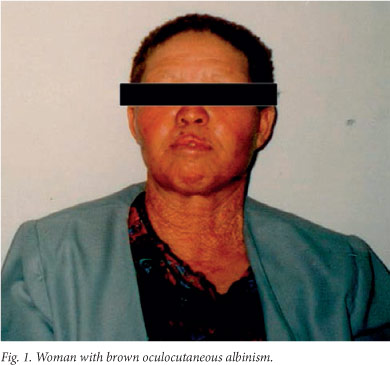
OCA3
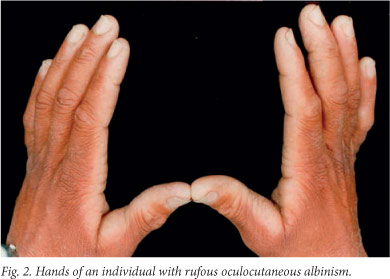
OCA3 (also known as rufous OCA) results from autosomal recessive mutations at the TYRP1 locus, at least in patients of African descent. Like OCA2, the precise function of this gene is not known, however, pigment production is significantly reduced in its absence. The prevalence of OCA3 among southern African blacks is at least 1/8 580, with a carrier rate of approximately 1/46,[30] while it is extremely rare in the remainder of the world. Witkop et at.[31] first described the characteristics of rufous albinism as 'mahogany red to deep red' hair, reddish-brown skin, occasional presence of pigmented nevi or freckles, reddish-brown to brown eye colour, slight transillumination of the iris, fundal pigment, nystagmus, photophobia and approximately 20/100 visual acuity (Fig. 2). Susceptibility to solar damage and skin neoplasia is lower than for OCA2 and brown OCA individuals. Affected individuals showed no evidence of photoaging, photodamage or carcinomas.[30] Our research identified two mutations that result in premature stop codons[29] and account for 90% of the mutated TYRP1 alleles in southern African individuals with OCA3. Both are nonsense mutations and there is unlikely to be any residual enzyme activity. Unlike normal black skin where the melanocytes contain mostly mature eumelanosomes, melanocytes in OCA3 skin contain both eumelanosomes and phaeomelanosomes at various stages of melanisation. Many of the organelles are, however, aberrant, 'crescent'- or racquet-shaped and have melanin only at the edges.[32]
Studies in mouse models have shown that TYRP1, also contributes to tyrosinase maturation. In melanocytes lacking TYRP1, tyrosinase accumulates in the ER,[33] though to a lesser degree than in OCA2-null melanocytes. TYRP1 has also been shown to stabilise tyrosinase,[34] leading to the suggestion that TYRP1 is required for transfer of tyrosinase from the ER to the Golgi.[35]
Genetic testing for OCA
Identification of the genes involved in these pigmentation disorders has facilitated the development of prenatal testing. Several groups have demonstrated the utility of genetic testing for albinism and five prenatal diagnoses have been carried out in the Human Genetics laboratory at the National Health Laboratory Service and University of the Witwatersrand, Johannesburg (F Essop, personal communication). Given that one mutation accounts for the majority of albinism in southern Africa, prenatal diagnosis may be a feasible option until effective therapies become available.
Vitiligo
Another type of pigmentation disorder common in southern Africa is vitiligo. Vitiligo affects 1 - 2% of people worldwide, occurring with similar frequencies in all ethnic groups. Phenotypically, it is characterised by acquired depigmented patches of skin resulting from the death of melanocytes. Various forms, defined by the distribution of the depigmented lesions, have been identified. These include generalised vitiligo (vitiligo vulgaris), the most common form with widely distributed, symmetric and progressive lesions; and segmental vitiligo, which presents with unilateral depigmented patches. The focus of this part of the review will be generalised vitiligo (referred to hereinafter as vitiligo).
Similar to OCA, vitiligo has a major impact on the physical and mental health of patients. Melanocyte loss reduces melanin-derived photoprotection of the skin and can compromise cutaneous immunity. Ocular melanocytes are occasionally lost, causing photophobia and night blindness.[36] Depigmentation commonly affects visible areas such as the face and hands, which has a significant impact on psychological well-being, especially among people of colour. Studies have shown that individuals with vitiligo are embarrassed by their appearance and usually feel uncomfortable socialising, leading to severe depression in some cases and, on rare occasions, suicide.[3] The early age of onset, typically in the first two decades of life, exacerbates the negative impacts of this disorder, preventing individuals from finding employment and even partners.[37]
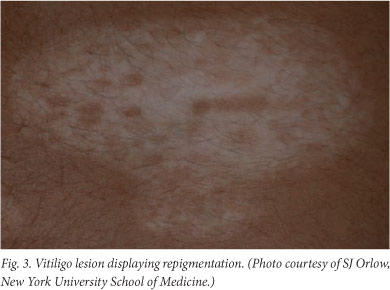
The pathophysiology of vitiligo is not well understood and there are few predictors of disease progression. While some individuals experience extensive pigment loss that may eventually affect the entire body, others undergo periodic changes in lesion size and number. Spontaneous repigmentation due to melanocyte migration from perilesional skin or unaffected hair follicles into areas of loss has been observed (Fig. 3). Treatments range from the use of topical corticosteroids and calcineurin inhibitors to UV combination treatments and, in some cases, skin grafting. In addition to potential side-effects such as skin atrophy, hyperpigmentation and scarring, these therapies are not effective in all patients and may have limited long-term results. The lack of new therapies is primarily due to the lack of clarity with regard to the aetiology of vitiligo.
Aetiology
Vitiligo is a multifactorial, non-Mendelian disorder which is associated with multiple loci. Vitiligo is thought to occur when melanocytes are unable to combat the stress induced by an environmental trigger such as severe sunburn, stress, vaccination, radiotherapy or exposure to cytotoxins.[38] In individuals genetically susceptible to developing vitiligo, the trigger event results in melanocyte dysfunction or death that, in turn, initiates an immune response which causes the spread of melanocyte loss. While vitiligo does not result in inflammation on the scale of skin disorders such as psoriasis, 'microinflammation' may also contribute to the onset of autoimmunity.[39]
A genetic component to vitiligo has long been suspected, due to the fact that incidence is higher among family members of affected individuals than in the general population. Recent genome-wide association studies have begun to identify the genetic factors that determine susceptibility to vitiligo.[40]
While the factors that initiate the onset of vitiligo are unclear at present, autoimmunity appears to be the mechanism by which lesions spread to multiple locations. The presence of antibodies targeting melanocyte-specific proteins in sera from patients has been reported on multiple occasions. Furthermore, 20 - 25% of patients with vitiligo have another autoimmune disorder such as psoriasis, diabetes and rheumatoid arthritis.[41] A number of immune-related genes have been found to associate with increased risk of vitiligo, including major histocompatibility class (MHC) I molecules as well as genes involved in autoimmune disorders, such as diabetes (UBASH3A and PTPN22) and rheumatoid arthritis (C1QTNF6).[40] In addition, melanocyte-specific genes have also been associated with increased risk, including OCA2, MC1R and TYR, which are thought to be the source of melanocyte-specific antigens.[42]
Oxidative stress and vitiligo
A constant factor in many hypotheses regarding the pathobiology of vitiligo is the involvement of oxidative stress. Studies have suggested that individuals with vitiligo have a compromised antioxidant response,[43] with enzymes such as catalase and superoxide dismutase present at higher than expected levels in tissue from perilesional areas and in sera from patients with vitiligo. [44]
Melanocytes in perilesional sites of vitiligo patients display hallmark dilation of the ER.[45] The ER is a sensor of cellular stress. Furthermore, protein maturation, which occurs primarily in the ER, requires a tightly regulated environment that allows for chemical bond formation which determines secondary and tertiary protein structure. Thus, disruption of the ER redox balance following cellular oxidative stress results in misfolded proteins, which in turn activates a stress pathway known as the unfolded protein response (UPR). The UPR ameliorates stress caused by accumulation of misfolded proteins in the ER by signalling a transient halt in global protein synthesis, increasing expression of chaperones that facilitate protein folding and increasing degradation of misfolded proteins.[46] The Manga laboratory has shown that chemical agents that trigger vitiligo induce oxidative stress that activates the UPR, suggesting a role for this pathway in the pathogenesis of vitiligo.[47] Interestingly, the UPR is activated in other autoimmune disorders such as type I diabetes[48] and contributes to activation of the immune response.
UPR activation in melanocytes results in increased expression of cytokines, such as interleukin (IL)-6 and -8 that can attract immune components to the skin which initiate an autoimmune response. [47] These cytokines have been found previously to be expressed at higher levels in perilesional compared with normal skin in vitiligo patients, suggesting that they may indeed play a role in the aetiology of vitiligo. This study thus identified a key link between events that trigger vitiligo and the onset of autoimmune disease.
Conclusion
Decades of work on albinism in SA, influenced by the views and contributions of Trefor Jenkins, his students and co-workers, has contributed to understanding the social and cultural milieu of albinism, the medical risks and implications, and unravelling the molecular basis and aetiology for OCA2 on the sub-continent (Table 1). The demystification of albinism and work with the Albinism Society of South Africa (http://www.albinism.org.za/), under the direction of Nomisonto Masibuko, has contributed to acceptance of the condition and empowerment to deal with this manageable trait to minimise the adverse effects on the lives of those with albinism.
As our understanding of the molecular basis of pigmentation improves, effective and long-lasting therapies are likely to become a reality for individuals with pigmentary disorders. Demonstrating that tyrosinase maturation and trafficking is key to multiple forms of OCA may make it feasible to develop a single approach for the treatment of multiple forms of OCA, while the identification of cytokines involved in the onset of vitiligo may facilitate the development of targeted therapies. Chaperones that promote protein folding are currently being developed for the treatment of Fabry's disease (Amicus Therapeutics), which results from misfolding of the enzyme α-galactosidase A, and antibodies targeting IL-6 are being tested for use in the treatment of non-small cell lung cancer.[49] Thus, therapies for OCA and vitiligo may now become a reality.
References
1. Gilchrest BA. Skin aging and photoaging: An overview. J Am Acad Dermatol 1989;21(3 Pt 2):610-613. [http://dx.doi.org/10.1016/S0190-9622(89)70227-9] [ Links ]
2. Hatchome N, Aiba S, Kato T, Torinuki W, Tagami H. Possible functional impairment of Langerhans' cells in vitiliginous skin. Reduced ability to elicit dinitrochlorobenzene contact sensitivity reaction and decreased stimulatory effect in the allogeneic mixed skin cell lymphocyte culture reaction. Arch Dermatol 1987;123(1):51-54. [http://dx.doi.org/10.1001/archderm.1987.01660250057017] [ Links ]
3. Porter J, Beuf AH, Lerner A, Nordlund J. Response to cosmetic disfigurement: Patients with vitiligo. Cutis 1987;39(6):493-494. [ Links ]
4. Kawakami A, Fisher DE. Key discoveries in melanocyte development. J Invest Dermatol 2011;131(E1):E2-E4. [http://dx.doi.org/10.1038/skinbio.2011.2] [ Links ]
5. Tassabehji M, Newton VE, Read AP. Waardenburg syndrome type 2 caused by mutations in the human microphthalmia (MITF) gene. Nat Genet 1994;8(3):251-255. [http://dx.doi.org/10.1038/ng1194-251] [ Links ]
6. Wei ML. Hermansky-Pudlak syndrome: A disease of protein trafficking and organelle function. Pigment Cell Res 2006;19(1):19-42. [http://dx.doi.org/10.1111/j.1600-0749.2005.00289.x] [ Links ]
7. Ebanks JP, Koshoffer A, Wickett RR, et al. Epidermal keratinocytes from light vs. dark skin exhibit differential degradation of melanosomes. J Invest Dermatol 2011;131(6):1226-1233. [http://dx.doi.org/10.1038/jid.2011.22] [ Links ]
8. Thong H-Y, Jee S-H, Sun C-C, Boissy RE. The pattern and mechanism of melanosome distribution in keratinocytes of human skin as one determining factor of skin colour. Br J Dermatol 2003;149:498- 505. [ Links ]
9. Nan H, Kraft P, Qureshi AA, et al. Genome-wide association study of tanning phenotype in a population of European ancestry. J Invest Dermatol 2009;129(9):2250-2257. [http://dx.doi.org/10.1038/jid.2009.62] [ Links ]
10. Rees JL. Genetics of hair and skin color. Annu Rev Genet 2003;37:67-90. [ Links ]
11. Sturm RA, Frudakis TN. Eye colour: Portals into pigmentation genes and ancestry. Trends Genet 2004;20(8):327-332. [http://dx.doi.org/10.1016/j.tig.2004.06.010] [ Links ]
12. Oetting WS, King RA. Molecular basis of type I (tyrosinase-related) oculocutaneous albinism: Mutations and polymorphisms of the human tyrosinase gene. Hum Mutat 1993;2(1):1-6. [http://dx.doi.org/10.1002/humu.1380020102] [ Links ]
13. Lee ST, Nicholls RD, Bundey S, Laxova R, Musarella M, Spritz RA. Mutations of the P gene in oculocutaneous albinism, ocular albinism, and Prader-Willi syndrome plus albinism. N Engl J Med 1994;330(8):529-534. [http://dx.doi.org/10.1056/NEJM199402243300803] [ Links ]
14. Manga P, Kromberg JG, Box NF, Sturm RA, Jenkins T, Ramsay M. Rufous oculocutaneous albinism in southern African blacks is caused by mutations in the TYRP1 gene. Am J Hum Genet 1997;61(5):1095-1101. [http://dx.doi.org/10.1086/301603] [ Links ]
15. Newton JM, Cohen-Barak O, Hagiwara N, et al. Mutations in the human orthologue of the mouse underwhite gene (uw) underlie a new form of oculocutaneous albinism, OCA4. Am J Hum Genet 2001;69(5):981-988. [http://dx.doi.org/10.1086/324340] [ Links ]
16. Kromberg JG, Zwane EM, Jenkins T. The response of black mothers to the birth of an albino infant. Am J Dis Child 1987;141(8):911-916. [ Links ]
17. Kromberg JG, Jenkins T. Albinism in the South African Negro. III. Genetic counselling issues. J Biosoc Sci 1984;16(1):99-108. [http://dx.doi.org/10.1017/S0021932000014838] [ Links ]
18. Ezeilo BN. Psychological aspects of albinism: An exploratory study with Nigerian (Igbo) albino subjects. Soc Sci Med 1989;29(9):1129-1131. [http://dx.doi.org/10.1016/0277-9536(89)90026-9] [ Links ]
19. Garrod AE. The Croonian lectures on inborn errors of metabolism. Lancet 1908;172(4427):73. [http://dx.doi.org/10.1016/S0140-6736(01)78482-6] [ Links ]
20. Stevens G, van Beukering J, Jenkins T, Ramsay M. An intragenic deletion of the P gene is the common mutation causing tyrosinase-positive oculocutaneous albinism in southern African Negroids. Am J Hum Genet 1995;56(3):586-591. [ Links ]
21. Kromberg JG, Jenkins T. Prevalence of albinism in the South African negro. S Afr Med J 1982;61(11):383-386. [ Links ]
22. Witkop CJ, Jr. Inherited disorders of pigmentation. Clin Dermatol 1985;3(1):70-134. [ Links ]
23. Onojafe IF, Adams DR, Simeonov DR, et al. Nitisinone improves eye and skin pigmentation defects in a mouse model of oculocutaneous albinism. J Clin Invest 2011;121(10):3914-3923. [http://dx.doi.org/10.1172/JCI59372] [ Links ]
24. Vanclooster A, Devlieger R, Meersseman W, et al. Pregnancy during nitisinone treatment for tyrosinaemia type I: First human experience. JIMD Rep 2012;5:27-33. [http://dx.doi.org/10.1007/8904_2011_88] [ Links ]
25. Gardner JM, Nakatsu Y, Gondo Y, et al. The mouse pink-eyed dilution gene: Association with human Prader-Willi and Angelman syndromes. Science 1992;257(5073):1121-1124. [http://dx.doi.org/10.1126/science.257.5073.1121] [ Links ]
26. Ramsay M, Colman MA, Stevens G, et al. The tyrosinase-positive oculocutaneous albinism locus maps to chromosome 15q11.2-q12. Am J Hum Genet 1992;51(4):879-884. [ Links ]
27. Kerr R, Stevens G, Manga P, et al. Identification of P gene mutations in individuals with oculocutaneous albinism in sub-Saharan Africa. Hum Mutat 2000;15(2):166-172. [http://dx.doi.org/10.1002/(SICI)1098-1004(200002)15:2<166::AID-HUMU5>3.3.CO;2-Q] [ Links ]
28. King RA, Creel D, Cervenka J, Okoro AN, Witkop CJ. Albinism in Nigeria with delineation of new recessive oculocutaneous type. Clin Genet 1980;17(4):259-270. [http://dx.doi.org/10.1111/j.1399-0004.1980.tb00145.x] [ Links ]
29. Manga P, Kromberg JG, Turner A, Jenkins T, Ramsay M. In southern Africa, brown oculocutaneous albinism (BOCA) maps to the OCA2 locus on chromosome 15q: P-gene mutations identified. Am J Hum Genet. 2001;68(3):782-787. [http://dx.doi.org/10.1086/318800] [ Links ]
30. Kromberg JG, Castle DJ, Zwane EM, et al. Red or rufous albinism in southern Africa. Ophthalmic Paediatr Genet 1990;11(3):229-235. [http://dx.doi.org/10.3109/13816819009020984] [ Links ]
31. Witkop CJJ, Quevedo WCJ, Fitzpatrick TB. Albinism and other disorders of pigment metabolism. In: Stanbury J, Wyngerden JB, Fredrickson DS, Goldstein JL, Brown MS, eds. The metabolic basis of inherited disease. New York: McGraw Hill,1983:301-346. [ Links ]
32. Kidson SH, Richards PD, Rawoot F, Kromberg JG. An ultrastructural study of melanocytes and melanosomes in the skin and hair bulbs of rufous albinos. Pigment Cell Res 1993;6(4 Pt 1):209-214. [http://dx.doi.org/10.1111/j.1600-0749.1993.tb00604.x] [ Links ]
33. Toyofuku K, Wada I, Valencia JC, Kushimoto T, Ferrans VJ, Hearing VJ. Oculocutaneous albinism types 1 and 3 are ER retention diseases: Mutation of tyrosinase or Tyrp1 can affect the processing of both mutant and wild-type proteins. Faseb J 2001;15(12):2149-2161. [http://dx.doi.org/10.1096/fj.01-0216com] [ Links ]
34. Manga P, Sato K, Ye L, Beermann F, Lamoreux ML, Orlow SJ. Mutational analysis of the modulation of tyrosinase by tyrosinase-related proteins 1 and 2 in vitro. Pigment Cell Res 2000;13(5):364-374. [http://dx.doi.org/10.1034/j.1600-0749.2000.130510.x] [ Links ]
35. Manga P, Orlow SJ. Informed reasoning: Repositioning of nitisinone to treat oculocutaneous albinism. J Clin Invest 2011;121(10):3828-3831. [http://dx.doi.org/10.1172/JCI59763] [ Links ]
36. Boissy R. Extracutaneous melanocytes. In: Nordlund J, Boissy R, Hearing V, King R, Ortonne J, eds. The Pigmentary System Physiology and Pathophysiology. New York: Oxford University Press, 1998:59-73. [ Links ]
37. Parsad D, Dogra S, Kanwar AJ. Quality of life in patients with vitiligo. Health Qual Life Outcomes 2003;1:58. [http://dx.doi.org/10.1186/1477-7525-1-58] [ Links ]
38. Nancy AL, Yehuda S. Prediction and prevention of autoimmune skin disorders. Arch Dermatol Res 2009;301(1):57-64. [http://dx.doi.org/10.1007/s00403-008-0889-3] [ Links ]
39. Taieb A. Vitiligo as an inflammatory skin disorder: A therapeutic perspective. Pigment Cell Melanoma Res 2012;25(1):9-13. [http://dx.doi.org/10.1111/j.1755-148X.2011.00939.x] [ Links ]
40. Jin Y, Birlea SA, Fain PR, et al. Variant of TYR and autoimmunity susceptibility loci in generalized vitiligo. N Engl J Med 2010;362(18):1686-1697. [http://dx.doi.org/10.1056/NEJMoa0908547] [ Links ]
41. Halder RM, Chappell JL. Vitiligo update. Semin Cutan Med Surg 2009;28(2):86-92. [http://dx.doi.org/10.1016/j.sder.2009.04.008] [ Links ]
42. Jin Y, Birlea SA, Fain PR, et al. Genome-wide association analyses identify 13 new susceptibility loc i for generalized vitiligo. Nat Genet 2012;44(676-680). [http://dx.doi.org/10.1038/ng.2272] [ Links ]
43. Yildirim M, Baysal V, Inaloz HS, Kesici D, Delibas N. The role of oxidants and antioxidants in generalized vitiligo. J Dermatol 2003;30(2):104-108. [ Links ]
44. Dammak I, Boudaya S, Ben Abdallah F, Turki H, Attia H, Hentati B. Antioxidant enzymes and lipid peroxidation at the tissue level in patients with stable and active vitiligo. Int J Dermatol 2009;48(5):476-480. [http://dx.doi.org/10.1111/j.1365-4632.2009.03998.x] [ Links ]
45. Le Poole C, Boissy RE. Vitiligo. Semin Cutan Med Surg 1997;16(1):3-14. [ Links ]
46. Malhotra JD, Kaufman RJ. The endoplasmic reticulum and the unfolded protein response. Semin Cell Dev Biol 2007;18(6):716-731. [http://dx.doi.org/10.1016/j.semcdb.2007.09.003] [ Links ]
47. Toosi S, Orlow SJ, Manga P. Vitiligo-inducing phenols activate the unfolded protein response in melanocytes resulting in upregulation of IL6 and IL8. J Invest Dermatol 2012;132(11):2601-2609. [http://dx.doi.org/10.1038/jid.2012.181] [ Links ]
48. Lipson KL, Fonseca SG, Ishigaki S, et al. Regulation of insulin biosynthesis in pancreatic beta cells by an endoplasmic reticulum-resident protein kinase IRE1. Cell Metab 2006;4(3):245-254. [http://dx.doi.org/10.1016/j.cmet.2006.07.007] [ Links ]
49. Bayliss TJ, Smith JT, Schuster M, Dragnev KH, Rigas JR. A humanized anti-IL-6 antibody (ALD518) in non-small cell lung cancer. Expert Opin Biol Ther 2011;11(12):1663-1668. [http://dx.doi.org/10.1517/14712598.2011.627850] [ Links ]
 Correspondence:
Correspondence:
P Manga
(prashiela.manga@nyumc.org)
Accepted 25 July 2013.

