










Serviços Personalizados
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkSAMJ: South African Medical Journal
versão On-line ISSN 2078-5135
versão impressa ISSN 0256-9574
SAMJ, S. Afr. med. j. vol.103 no.12 Pretoria Dez. 2013
RESEARCH
Combined paediatric liver-kidney transplantation: Analysis of our experience and literature review
B StrobeleI; J LovelandII, III; R BritzIV; E GottlichV; A WelthagenVI; J BothaVII
IMB ChB, FCS (SA). Department of Surgery, University of the Witwatersrand, Johannesburg, South Africa
IIMB ChB, FCS (SA), Cert Paed Surg. Transplant Division, Wits Donald Gordon Medical Centre, University of the Witwatersrand, Johannesburg, South Africa
IIIMB ChB, FCS (SA), Cert Paed Surg. Department of Paediatric Surgery, Chris Hani Baragwanath Academic Hospital, University of the Witwatersrand, Johannesburg, South Africa
IVMB BCh, DA (SA), FCS (SA). Transplant Division, Wits Donald Gordon Medical Centre, University of the Witwatersrand, Johannesburg, South Africa
VMB BCh, DCH, FCP Paed (SA), Cert Nephrology Paed. Transplant Division, Wits Donald Gordon Medical Centre, University of the Witwatersrand, Johannesburg, South Africa
VIMB BCh, FCS (SA). Department of Paediatric Surgery, Chris Hani Baragwanath Academic Hospital, University of the Witwatersrand, Johannesburg, South Africa
VIIMB BCh, FCS (SA). Transplant Division, Wits Donald Gordon Medical Centre, University of the Witwatersrand, Johannesburg, South Africa
ABSTRACT
BACKGROUND: Renal insufficiency is increasingly common in end-stage liver disease and allocation of livers to this category of patient has escalated. The frequency of combined liver-kidney transplantation (CLKT) has consequently increased. Indications for CLKT in children differ from those for adults and typically include rare congenital conditions; subsequently limited numbers of this procedure have been performed in paediatric patients worldwide. Scant literature exists on the subject.
METHODS: Subsequent to institutional approval, a retrospective chart analysis of all paediatric CLKTs performed at the Transplant Unit, Wits Donald Gordon Medical Centre, University of the Witwatersrand, Johannesburg, South Africa between January 2005 and July 2013 was conducted.
RESULTS: Defining children as younger than 18 years of age, 43 patients had received a liver transplant since 2005, of whom 8 received a CLKT. Indications included autosomal recessive polycystic kidney disease (n=3), primary hyperoxaluria type 1 (n=4) and heterozygous factor H deficiency with atypical haemolytic uraemic syndrome (n=1). Graft combinations included whole liver and one kidney (n=5), whole liver and two kidneys (n=1) and left lateral liver segment and one kidney (n=2), all from deceased donors. Patient age ranged from 4 to 17 years (median 9) and included 4 females and 4 males. Weight ranged from 13 to 42 kg (median 22.5). We describe one in-hospital mortality. The remaining 7 patients were long-term survivors with a survival range from 6 to 65 months.
CONCLUSIONS: Although rarely indicated in children, CLKT is an effective treatment option, appropriately utilising a scarce resource and significantly improving quality of life in the recipient.
The Transplant Unit, Wits Donald Gordon Medical Centre, University of the Witwatersrand, Johannesburg, South Africa has performed 43 paediatric liver transplants to date. Of these, 8 were combined liver-kidney transplants. Since the first successful kidney and liver transplants in the 1950s and 1960s respectively, both patient and graft survival have improved dramatically.[1] The first successful combined liver-kidney transplantation (CLKT) was subsequently performed by Margreiter in Austria in 1984.[2] Renal insufficiency, secondary to a variety of causes, is a common problem in patients suffering from endstage liver disease, particularly in adults.[3] Renal impairment in liver transplant patients amplifies the risk for both postoperative chronic kidney disease (CKD) and procedure-related mortality. With the introduction of the Model for End-Stage Liver Disease (MELD) score for liver transplantation the allocation of organs to patients with renal insufficiency has increased.[1-3] Consequently, the number of CLKTs in adults has shown a considerable increase over the last few years. In children, the most common causes for CLKT are congenital diseases affecting both liver and kidney, such as primary hyperoxaluria type 1 (PH1) and autosomal recessive polycystic kidney disease (ARPKD). [2] These diseases have a low incidence and consequently CLKT in children remains a therapy performed in a very limited number of cases with little data available in the literature.[1]
Methods
All paediatric patients undergoing CLKT were identified retrospectively. Eight transplants were performed in 8 children (<18 years of age on the day of transplant) since 2005. Recipient characteristics are listed in Table 1. Pertinent intra- and postoperative events were documented, as were any postoperative complications (particularly those related to vascular, biliary or urinary reconstruction) and graft and patient survival were analysed. Finally, a retrospective, English-language PubMed search was conducted in January 2013 for all published papers relating to CLKT.

Results
Eight CLKTs were performed, none of the children having received previous transplants. Indications included congenital hepatic fibrosis associated with polycystic kidney disease (PKD) (n=3), PH1 (n=4) and heterozygous factor H deficiency with atypical haemolytic uraemic syndrome (aHUS) (n=1). Patients ranged in age from 4 to 17 years (median 9) and included 4 females and 4 males. Weight ranged from 13 to 42 kg (median 22.5). Waiting times on the active transplant list ranged from 49 to 1 383 days (median 270). The range of post-transplant follow-up was 6 - 65 months.
Graft combinations were as follows: whole liver and one kidney (n=5); whole liver and two kidneys (n=1) and left lateral segment of liver and one kidney (n=2). Median anaesthetic time was 7 hrs and blood loss ranged from 80 to 4 970 ml (median 1 831). Median blood volume transfused was 900 ml of leukocyte-depleted packed red blood cells and other blood products, and 450 ml of cell-saved whole blood. Blood group crossmatch was identical in 7 patients and compatible in 1 patient.
Induction immunosuppression consisted of daclizumab (Zenapax) (weight <40 kg: 10 mg intravenous injection (IVI) on days 1 and 4; weight >40 kg: 20 mg IVI on days 1 and 4) and methylprednisolone (Solu-Medrol) (600 mg/m2 to a maximum of 500 mg). Postoperative maintenance immunosuppression consisted of mycophenolate mofetil (CellCept), tacrolimus (Prograf) and prednisone. In one case, everolimus was used after conversion from the calcineurin inhibitor. With respect to preoperative dialysis, 6 patients received haemodialysis, 1 peritoneal dialysis, and 1 was transplanted pre-emptively. The child with aHUS received polyvalent human normal immunoglobulin (Polygam) and plasma exchange pre-transplant, as well as for 10 days post-transplant. Eculizumab was not used as it was not available in South Africa at the time. No patients required postoperative dialysis for delayed graft function. All patients with hyperoxaluria received 3 months of postoperative haemodialysis to decrease systemic oxalate load. This patient group presented pre-transplant with manifestations of systemic oxalosis, particularly dense nephrocalcinosis. One patient underwent sequential nephrectomies, the first pre-and the second post-CLKT, both for severe nephrocalcinosis and recurrent urinary tract infections. A second patient is currently being considered for bilateral nephrectomy for the same reason. Native kidneys in the remaining patients remained asymptomatic and were left in situ.
There was one mortality in this series in a patient with ARPKD. The remaining 7 patients were dialysis free, with excellent quality of life subjectively, and a mean follow-up of 2.17 years. The mortality was related to surgical complications of hepatic artery and portal vein thrombosis peri-operatively. Although the patient was re-listed, a second liver did not become available and the patient died of acute hepatic failure. In the patient who received the two kidneys as an en bloc graft from a 2-year-old donor, one kidney infarcted secondary to renal artery thrombosis, although this had no impact on renal function. Another patient developed a wound haematoma, which resolved with conservative management. The last patient, who received a reduced left lateral segment graft and single kidney, developed abdominal compartment syndrome that required urgent decompression and closure with a large mesh.
Three of the patients presented with mild episodes of rejection. The first had a biochemically diagnosed episode of mild liver rejection on day 15 post transplant. This was successfully managed with 3 doses of methylprednisolone without biopsy. The second patient presented with a histologically confirmed episode of liver rejection two years post transplant. This also settled completely after increasing the tacrolimus dose and adding methylprednisolone. The third patient, with aHUS, was transferred to another centre for long-term follow-up. Presenting with a clinically suggestive episode of mild liver rejection, despite a normal liver biopsy, the patient was successfully treated with methylprednisolone and increased dose of tacrolimus. Currently all 7 patients have excellent hepatic and renal function. We could not conclusively define any documentable immunological protection to the renal graft in this small patient cohort.
Technical details of the CLKT
The specific surgical techniques for organ procurement, graft preparation and the respective implant procedures are well described. All kidneys were placed extra-peritoneally via a right-sided RutherfordMorrison incision. We did not encounter the theoretical risk of skin bridge necrosis at the lateral aspects of the right subcostal and Rutherford-Morrison incisions where the two approximate. The right-sided approach allows excellent access to the distal aorta and inferior vena cava, which may be required to establish adequate arterial inflow and venous outflow, respectively. This is especially important when a large donor kidney is implanted into a small child (weighing <20 kg), when up to 30% of the circulating blood volume is taken up by the transplanted kidney on release of clamps. Inadequate arterial inflow will compromise perfusion, causing subsequent necrosis of the graft. In this situation arterial inflow to the donor kidney is established directly off the aorta, below the origin of the inferior mesenteric artery. This approach was utilised in 3/8 renal grafts.
Discussion
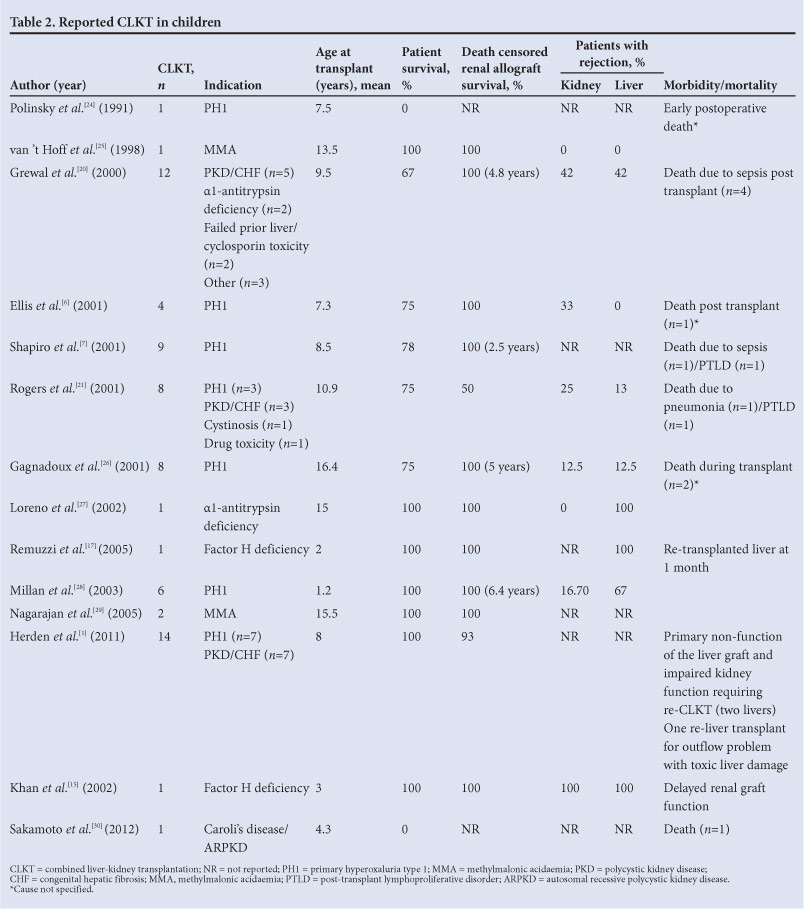
All published paediatric CLKTs extracted from the PubMed database are shown in Table 2. While these total 101 cases, 3 568 cases have been reported from 1984 to the present. The United Network for Organ Sharing (UNOS), however, reports that 231 CKLTs have been performed in children in 41 transplant centres.[5] The median number of transplants per centre was 3 (average 6). The majority of transplants were for PH1 (n=52, 48.6%), ARPKD (n=20, 18.5%) and atypical factor H deficiency (n=8, 7.4%). Immunosuppression varied significantly from tacrolimus or cyclosporin A monotherapy to multiple drug combinations and even quadruple therapy with antithymoglobulin, cyclosporin A, methylprednisolone and azathioprine. When reported, complications were predominantly sepsis related (n=6, 5.94%), and invariably resulted in mortality. The relative infrequency of CLKT in children is as a result of limited indications, which include rare conditions with low incidences that have not changed over the years.[2] This leaves a challenging problem in determining the optimal indications for CLKT. The indications for CLKT in our patient cohort are listed in Table 1. It is evident, even with the small numbers that we are reporting on, that our indications and numbers mirror those of the world literature. We will discuss the three most common indications for CLKT, namely PH1, ARPKD with congenital hepatic fibrosis and heterozygous factor H deficiency with aHUS.
PH1 is an autosomal recessive metabolic condition caused by a congenital deficiency of alanine-glyoxylate aminotransferase (AGT), a hepatic peroxisomal enzyme. Patients with this deficiency metabolise glyoxylate to oxalate and glycolic acid instead of glycine.[6] The kidneys are subsequently burdened with eliminating the large oxalate load, which results in deposition of calcium-oxalate crystals in the kidney and progression to chronic renal failure. The condition is often more severe in infants, with end-stage renal disease (ESRD) presenting before 2 years of age. By 15 years of age 50% of patients will have developed ESRD.[1,3,7] As the glomerular filtration rate declines, oxalate is deposited systemically into bone and soft tissues including the retina, blood vessels, nerves and heart.[6] If the condition is diagnosed early, and before significant renal failure is established, isolated liver transplantation (iLT) is an acceptable option, with excellent survival.[7] However, this only addresses the issue of oxalate formation and does not resolve the problem of an already high burden of oxalate, which would force the kidneys to eliminate the remaining stores. This can contribute to progressive CKD requiring a renal transplant at a later stage. The converse is, however, not acceptable - graft survival after isolated kidney transplantation (iKT) for PH1 approximates 18% at 3 years, based on data from the European Dialysis and Transplant Association registry.[8] This obviously occurs due to progressive nephrocalcinosis and urolithiasis and early graft failure, as hepatic AGT is still deficient, and oxalate over-production continues.
CLKT addresses both the primary metabolic insult, namely hepatic AGT deficiency, and the consequent end-organ damage secondary to oxalosis, i.e. chronic renal failure. Reported outcomes have been varied. The largest series of patients is from the European PH1 Transplant Registry; 127 transplants were performed in 117 patients between 1984 and 2005, 99 CLKTs, 22 iLTs and 6 patients underwent iLT followed by iKT. The 10-year survival was 69% for all groups.[9] Factors which were clearly shown to affect outcome and improve survival were the overall clinical condition of the patient at the time of transplantation, earlier diagnosis and aggressive pre-transplantation dialysis.[1,2,4,9] Of interest is that, despite a new liver, >50% of renal graft failures following CLKT were due to oxalate deposition in the new kidney and that hyperoxaluria was detectable up to 45 months post transplantation. This oxalate burden should not be underestimated.[2,10] One of our patients required both haemo-and peritoneal dialysis preoperatively. Our current protocol is to perform haemodialysis 4 - 5 times per week pre-transplant and for 3 months post transplantation to decrease the oxalate load and to mitigate any damage to the renal allograft by nephrocalcinosis or urolithiasis. Consideration should also be given to the shortage of deceased donor organs and the resulting pressure to other patients on the waiting list. Thus, alternatives that should be considered once the decision to list has been taken include the feasibility of related living kidney donation as and when a deceased donor liver becomes available, as well as consideration of both related living kidney and liver transplantation simultaneously.[1,11]
ARPKD has an incidence of 1/20 000 live births and demonstrates a variable clinical progression.[2,12] The most severely affected children present with ESRD, either in the perinatal period or during early infancy. Liver involvement is a result of congenital hepatic fibrosis or Caroli disease, which may progress to portal hypertension, hypersplenism with resultant thrombocytopenia and recurrent ascending cholangitis (Caroli syndrome).[6,13] As the liver disease is chronic and may not necessarily progress to failure, iKT has been the standard intervention for ESRD secondary to ARPKD with minimal liver disease. Reported data on iKT show patient survival to be 89% and death censored graft survival at 100% at 5 years.[14] However, Khan et al.[15] reported on 14 patients who had received iKT in the presence of hepatic disease; 4/5 deaths in this series were due to complications from hepatic disease (systemic sepsis secondary to cholangitis) and 56% of the remaining survivors had complications related to congenital hepatic fibrosis at a mean of 6.3 years post transplantation. Davis et al.[13] published a retrospective study of 203 patients who received iKT for similar indications. Of particular interest in this study is the 64% of sepsis-related deaths. While sepsis is acknowledged as a common cause of death among the paediatric group of kidney transplant recipients, the North American Pediatric Renal Trials and Collaborative Studies registry report this at 28.9%.[6,16] Although not proven, this may hypothetically suggest an increased rate of cholangitis related to congenital hepatic fibrosis, and the fact that these patients already have a heavily colonised biliary tree. This situation is possibly accentuated by subsequent immunosuppression.[7,13] While liver disease is always present in these patients, it does not always warrant transplantation and iKT is often all that is required. When there is liver disease causing progressive and severe complications, CLKT is advocated. Although limited, outcome data following both adult and paediatric CLKT are favourable in the literature. CLKT should be considered when ESRD (as a result of ARPKD) and severe congenital hepatic fibrosis or hepatic failure are present. On occasion, liver disease secondary to the congenital hepatic fibrosis dominates the clinical picture. However, we are not aware of any cases of iLT in the literature and CLKT is recommended currently in this scenario.[8,11]
Heterozygous factor H deficiency with aHUS is a very unusual indication for CLKT, with only 7 cases described in the literature to date. Factor H is produced in the liver and regulates the activation of the complement cascade via the alternative pathway. Qualitative or quantitative deficiencies in factor H can result in the uninhibited deposition of complement, destruction of microvasculature and phenotype of HUS. There are >60 heterozygous factor H mutations that are responsible for ~30% of recurrent cases of HUS. They carry a poor prognosis, resulting in ESRD or death in 50% of patients.[3,9,17,18] Outcomes following iKT are equally poor, with recurrences in the range of 50 - 100%.[3,17] CLKT addresses both the qualitative and quantitative factor H issues and restores renal function. Recently, eculizumab has been registered for the management of aHUS. Eculizumab is a recombinant, humanised monoclonal antibody targeting the complement protein C5. It protects red blood cells from chronic intravascular haemolysis by preventing generation of the C5b9 complex, thus inhibiting activation of terminal complement, which is responsible for cell lysis. The current recommendation is to perform a kidney transplant combined with lifelong eculizumab therapy.[19] Unfortunately, the lifelong cost of this drug is currently prohibitively high. The alternative is CLKT, which is a more aggressive treatment option and is potentially associated with greater morbidity and mortality due to the combined organ transplant. To reduce this risk, an option is to use one dose of eculizumab peri-operatively in CLKTs to reduce the complications related to uninhibited complement activation. A second dose can be held in reserve if necessary. Where antifactor H antibodies exist a CKLT may need to be combined with lifelong plasma exchange and therapy aimed at reducing antibody levels. Our patient did not have antifactor H antibodies.
At the time of writing, CLKT has been reported in 7 patients (including our own case) with factor H-associated HUS. Most recently, Khan et al.[15] reported one case that was uncomplicated, apart from a short period of delayed renal graft function of the kidney. Saland et al.[3] also published a single case with 100% dual graft survival at 2 years. Our case was equally uncomplicated, with dual graft survival >2 years post transplant. Plasma exchange has been suggested in the immediate pre-transplant period, or intraoperatively, as it restores normal factor H function and inhibits unregulated complement deposition until the new liver has had time to normalise factor H levels and function. Also, because split liver grafts are more susceptible to poor perfusion and the resultant increased complement activation, it is suggested by some units that whole liver grafts be used.[18] It should be stated that this is not the practice of our unit and we would consider the use of split grafts without reservation. Although there are only a limited number of documented cases in addition to our own single case experience, it has been demonstrated that, in light of these recent successes, as well as the poor outcomes of alternative therapy, CLKT should be considered for this subgroup of patients.
Since CLKTs are now more commonly performed, there has been debate as to whether the liver allograft offers 'immunoprotection' to the kidney allograft. In 2003, a review of the UNOS database illustrated a lower 6-month cumulative acute renal rejection rate for patients receiving CLKT than iKT (21.5% v. 30.1%, respectively). When reviewing single-centre experiences, the results appear even more promising; however, the small numbers reported in these studies should caution their interpretation. Grewal et al.[20] reported a 42% acute renal allograft rejection rate but only 2/5 cases were proven by biopsy. Rogers et al.[21] showed a 25% acute renal allograft rejection rate v. a 86% rejection rate in the kidney after liver transplant cohort. More recently, de la Cerda et al.[22] conducted a single-centre, retrospective, case-control study that looked at 10 children who had CLKT and survived to 6 months, comparing them to a control group of 20 kidney-only transplants matched for age, era and immunosuppression. In the CLKT group, only 1 acute renal allograft rejection was reported at 7 years secondary to non-compliance of medication. In the same period, the kidney-only group had 16 acute rejection episodes. There was also no hepatic rejection in the CLKT group. The study was, however, too small to uncover a significant difference in immunologically mediated kidney allograft failure. The same authors therefore looked at UNOS paediatric data, analysing 111 CLKT and 3 798 kidney-only transplants between 1995 and 2005. They found that, although renal graft loss in the first 6 months was higher in the CLKT than the kidney-only group (20.1% v. 5.9%, respectively), death-censored kidney allograft survival at 5 years was significantly better in the CLKT group and that function did not continue to deteriorate as it did in the kidney-only group.[22]
When reviewing the literature there are a few reported cases where patients required re-liver transplant after CLKT for graft non-function or rejection. In one case, a patient received two subsequent liver transplants following the initial CLKT.[23] Although that patient did not ultimately survive, the feasibility of subsequent transplantation does exist, dependent obviously on the availability of organs. This does validate our suggestion that our single mortality might have survived had another organ been available and, at the same time, raises the question of utilising a scarce resource.
Conclusion
Our centre demonstrates equivalent results in terms of graft survival, morbidity and mortality compared with reported data in the international literature. In the paediatric age group, the most common indications for CLKT are metabolic diseases, which affect either the kidney alone with or without liver dysfunction or disorders affecting both organs simultaneously. CLKT has good results in select groups of patients and long-term survival approaches that of liver transplantation alone. Outcomes are optimised by early evaluation and listing, before patients manifest systemic complications and develop end-stage disease. It is our strong view that all options should be utilised in transplanting these patients, including deceased donor split livers, RLD kidneys with deceased donor liver, and the RLD option for both liver and kidney. In patients with PH1, nephrocalcinosis and systemic oxalosis can be problematic post transplant and 3 months dialysis post CLKT to clear systemic oxalic acid is recommended. Irreversible renal dysfunction should not exclude children with severe liver disease from consideration for liver transplantation if it is performed with a simultaneous kidney transplant. Factors that negatively affect the outcome of iLT (UNOS status and re-transplantation) must be considered in determining the suitability of any given candidate for CLKT but ultimately ESRD and poor quality of life need to be offset against the potential excellent long-term benefits of a timeous CLKT. Similarly, patients with ARPKD demonstrate excellent graft and patient survival. Regarding mortality occurring in ARPKD, 64 - 80% can be attributed to systemic sepsis, likely a consequence of cholangitis subsequent to their liver disease. As surgical mortality in paediatric liver transplant recipients has been shown to be <10% at one year, CLKT is a viable option to decrease overall mortality in patients with recurrent cholangitis or complications of portal hypertension. There is suggestive clinical evidence that CLKT offers a degree of immunological protection to the renal allograft.
References
1. Herden U, Kemper M, Ganschow R, et al. Surgical aspects and outcome of combined liver and kidney transplantation in children. Transpl Int 2011;24(8):805-811. [http://dx.doi.org/10.1111/j.1432-2277.2011.01278.x] [ Links ]
2. Sutherland SM, Alexander SR, Sarwal MM, Berquist WE, Concepcion W. Combined liver-kidney transplantation in children: Indications and outcome. Pediatr Transplant 2008;12(8):835-846. [http://dx.doi.org/10.1111/j.1399-3046.2008.01041.x] [ Links ]
3. Saland JM, Emre SH, Shneider BL, et al. Favorable long-term outcome after liver-kidney transplant for recurrent hemolytic uremic syndrome associated with a factor H mutation. Am J Transplant 2006;6(8):1948-1952. [http://dx.doi.org/10.1111/j.1600-6143.2006.01375.x] [ Links ]
4. Chava SP, Singh B, Pal S, Dhawan A, Heaton ND. Indications for combined liver and kidney transplantation in children. Pediatr Transplant 2009;13(6):661-669. [http://dx.doi.org/10.1111/j.1399-3046.2008.01046.x] [ Links ]
5. UNOS Database. Combined Liver-Kidney Transplants by Age of Donor. http://optn.transplant.hrsa.gov/latestData/advancedData.asp (accessed 31 October 2013). [ Links ]
6. Ellis SR, Hulton SA, McKiernan PJ, de Ville, de Goyet J, Kelly DA. Combined liver-kidney transplantation for primary hyperoxaluria type 1 in young children. Nephrol Dial Transplant 2001;16(2):348-354. [http://dx.doi.org/10.1093/ndt/16.2.348] [ Links ]
7. Shapiro R, Weismann I, Mandel H, et al. Primary hyperoxaluria type 1: Improved outcome with timely liver transplantation: A single-center report of 36 children. Transplantation 2001;72(3):428-432. [http://dx.doi.org/10.1097/00007890-200108150-00012] [ Links ]
8. Broyer M, Brunner FP, Brynger H, et al. Kidney transplantation in primary oxalosis: Data from the EDTA registry. Nephrol Dial Transplant 1990;5(5):332-336. [http://dx.doi.org/10.1093/ndt/5.5.332] [ Links ]
9. Jamieson NV. A 20-year experience of combined liver/kidney transplantation for primary hyperoxaluria (PH1): The European PH1 Transplant registry experience 1984 - 2004. Am J Nephrol 2005;25(3):282-289. [http://dx.doi.org/10.1159/000086359] [ Links ]
10. Latta K, Jamieson NV, Scheinman JI, et al. Selection of transplantation procedures and perioperative management in primary hyperoxaluria type 1. Nephrol Dial Transplant 1995;10(Suppl 8):53-57. [http://dx.doi.org/10.1093/ndt/10.supp8.53] [ Links ]
11. Telega G, Cronin D, Avner ED. New approaches to the autosomal recessive polycystic kidney disease patient with dual kidney-liver complications. Pediatr Transplant 2013;17(4):328-335. [http://dx.doi.org/10.1111/petr.12076] [ Links ]
12. Zerres K, Mücher G, Becker J, et al. Prenatal diagnosis of autosomal recessive polycystic kidney disease (ARPKD): Molecular genetics, clinical experience, and fetal morphology. Am J Med Genet 1998;76(2):137-144. [http://dx.doi.org/10.1002/(SICI)1096-8628(19980305)76:2<137::AID-AJMG6>3.0.CO;2-Q] [ Links ]
13. Davis ID, Ho M, Hupertz V, Avner ED. Survival of childhood polycystic kidney disease following renal transplantation: The impact of advanced hepatobiliary disease. Pediatr Transplant 2003;7(5):364-369. [http://dx.doi.org/10.1034/j.1399-3046.2003.00094.x] [ Links ]
14. Beaunoyer M, Snehal M, Li L, Concepcion W, Salvatierra O, Sarwal M. Optimizing outcomes for neonatal ARPKD. Pediatr Transplant 2007;11(3):267-271. [http://dx.doi.org/10.1111/j.1399-3046.2006.00644.x] [ Links ]
15. Khan K, Schwarzenberg SJ, Sharp HL. Morbidity from congenital hepatic fibrosis after renal transplantation for autosomal recessive polycystic kidney disease. Am J Transplant 2002 [http://dx.doi.org/10.1034/j.1600-6143.2002.20412.x] [ Links ]
16. Smith JM, Stablein DM, Munoz R, Hebert D, McDonald RA. Contributions of the Transplant Registry: The 2006 Annual Report of the North American Pediatric Renal Trials and Collaborative Studies (NAPRTCS). Pediatr Transplant 2007;11(4):366-373. [http://dx.doi.org/10.1111/j.1399-3046.2007.00704.x] [ Links ]
17. Remuzzi G, Ruggenenti P, Colledan M, et al. Hemolytic uremic syndrome: A fatal outcome after kidney and liver transplantation performed to correct factor H gene mutation. Am J Transplant 2005;5(5):1146-1150. [http://dx.doi.org/10.1111/j.1600-6143.2005.00783.x] [ Links ]
18. Jalanko H, Peltonen S, Koskinen A, et al. Successful liver-kidney transplantation in two children with aHUS caused by a mutation in complement factor H. Am J Transplant 2008;8(1):216-221. [http://dx.doi.org/10.1111/j.1600-6143.2007.02029.x] [ Links ]
19. Connock M, Wang D, Fry-Smith A. Prevalence and Prognosis of Paroxysmal Nocturnal Haemoglobinuria and the Clinical and Cost-effectiveness of Eculizumab. Department of Public Health and Epidemiology. West Midlands Health Technology Assessment Collaboration Report; 69. Birmingham: University of Birmingham, 2008. [ Links ]
20. Grewal HP, Brady L, Cronin D, et al. Combined liver and kidney transplantation (lkt) in children. Transplantation 1998;66(8):50. [http://dx.doi.org/10.1097/00007890-199810270-00230] [ Links ]
21. Rogers J, Bueno J, Shapiro R, et al. Results of simultaneous and sequential pediatric liver and kidney transplantation. ransplantation 2001;72(10):1666-1670. [http://dx.doi.org/10.1097/00007890-200111270-00016] [ Links ]
22. de la Cerda F, Jimenez WA, Gjertson DW, Venick R, Tsai E, Ettenger R. Renal graft outcome after combined liver and kidney transplantation in children: UCLA and UNOS experience. Pediatr Transplant 2009;14(4):459-464. [http://dx.doi.org/10.1111/j.1399-3046.2009.01264.x] [ Links ]
23. Chava SP, Singh B, Stangou A, et al. Simultaneous combined liver and kidney transplantation: A single center experience. Clin Transplant 2009;24(3):E62-E68. [http://dx.doi.org/10.1111/j.1399-0012.2010.01168.x] [ Links ]
24. Polinsky MS, Dunn S, Kaiser BA, et al. Combined liver-kidney transplantation in a child with primary hyperoxaluria. Pediatr Nephrol 1991;5(3):332-334. [http://dx.doi.org/10.1007%2FBF00867495] [ Links ]
25. van't HoffWG, Dixon M, Tayolr J, et al. Combined liver-kidney transplantation in methylmalonic acidemia. J Pediatr 1998;132(6):1043-1044. [http://dx.doi.org/10.1016%2FS0022-3476%2898%2970407-X] [ Links ]
26. Gagnadoux MF, Lacaille F, Niaudet P, et al. Long term results of liver-kidney transplantation in children with primary hyperoxaluria. Pediatr Nephrol 2001;16(12):946-950. [http://dx.doi.org/10.1007%2Fs004670100001] [ Links ]
27. Loreno M, Boccagni P, Rigotti P, Naccarato R, Burra P. Combined liver-kidney transplantation in a 15-year-old boy with alpha1-antitrypsin deficiency. J Hepatol 2002;36(4):565-568. [http://dx.doi.org/10.1016%2FS0168-8278%2802%2900012-0] [ Links ]
28. Millan MT, Berquist WE, So SK, et al. One hundred percent patient and kidney allograft survival with simultaneous liver and kidney transplantation in infants with primary hyperoxaluria: A single-center experience. Transplantation 2003;76(10):1458-1463. [http://dx.doi.org/10.1097/01.TP.0000084203.76110.AC] [ Links ]
29. Nagarajan S, Enns GM, Millan MT, Winter S, Sarwal MM. Management of methylmalonic acidaemia by combined liver-kidney transplantation. J Inherit Metab Dis 2005;28(4):517-524. [http://dx.doi.org/10.1007/s10545-005-0517-8] [ Links ]
30. Sakamoto S, Kasahara M, Fukuda A, et al. Pediatric liver-kidney transplantation for hepatorenal fibrocystic disease from a living donor. Pediatr Transplant 2012;16(1):99-102. [http://dx.doi.org/10.1111%2Fj.1399-3046.2011.01609.x] [ Links ]
 Correspondence:
Correspondence:
J Loveland
(loveland@wol.co.za)
Accepted 7 October 2013


{kind=link}