










Servicios Personalizados
Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkSAMJ: South African Medical Journal
versión On-line ISSN 2078-5135
versión impresa ISSN 0256-9574
SAMJ, S. Afr. med. j. vol.103 no.9 Pretoria ene. 2013
GUIDELINE
Guideline for the diagnosis and management of multiple sclerosis: A Southern African perspective
D L GiampaoloI; BhigjeeII; C RetiefIII; M IsaacsI; M BritzIV; D OppermanV; R GovenderVI; M van RensburgIII
INetcare Rosebank Hospital, Johannesburg, South Africa
IIDepartment of Neurology, Inkosi Abert Luthuli Hospital, Durban, South Africa
IIILife Wilgers Hospital, Pretoria, South Africa
IVNetcare Greenacres Hospital, Port Elizabeth, South Africa
VConstantiaberg Medi-Clinic, Cape Town, South Africa
VIPaediatric Multiple Sclerosis Section, Ethekwini Hospital and Heart Centre, Durban, South Africa
ABSTRACT
Before making a diagnosis of multiple sclerosis (MS), it is imperative that alternative diagnoses are considered and excluded. This is particularly important in South Africa, which is a moderate prevalence MS area, has a high burden of neurological infections and where the majority of the people are black - an ethnic group that has a very low frequency of MS. Before applying diagnostic criteria, there should be no better explanation for the patient's presentation. This guideline, written on behalf of the Multiple Sclerosis Society of South Africa, aims to assist in the diagnosis and treatment of MS in Southern Africa.
1. Introduction
Multiple sclerosis (MS) is the most common neurological disorder to affect young adults in the developed world. It is characterised by an autoimmune process involving autoreactive B- and T-cells that results in demyelination, axonal loss and resultant gliosis.
It is frequently referred to as a white matter disease, but there is also cortical and deep grey matter involvement as noted with high Tesla magnetic resonance imaging (MRI) machines.
The specific cause still remains a mystery. However, it is thought to be a complex disease triggered in susceptible individuals by genes, epigenetics and environmental factors such as Epstein Barr virus, vitamin D and sunlight, and smoking, which are all well recognised epidemiological risk factors.
Although demyelination is the defining feature in a MS lesion, axonal loss is well described and occurs early on in the disease, making it necessary to treat patients as soon as possible. Axonal loss is thought to be mediated by cytotoxic T-cells, macrophages, antibodies, oxidative stress and loss of trophic support by oligodendrocytes.
Remyelination and repair has to occur in a proinflammatory auto-destructive environment, which has to change to a reparative one, thought to be initiated by interleukin (IL)-4, IL-10, brain-derived neurotrophic factor and transforming growth factor (TGF) beta. Defining these mechanisms that promote remyelination and devising appropriate trials is the key to developing effective treatments that decrease inflammation and, more importantly, promote repair.
1.1 Prevalence
The worldwide prevalence of MS is estimated at >2 million,[1,2] but there are considerable geographic and ethnic differences regarding the frequency of the disease. Previous reported prevalence rates vary from 140/100 000 in North-East Scotland, 50 - 100/100 000 in the Northern United States (US) and Canada to 4/100 000 in South America and sub-Saharan Africa.[2] The geographic variation has led to grading of prevalence in different parts of the world as high (>30/100 000), medium (5 - 29/100 000) or low (0 - 4/100 000). Ethnic differences are noted even in areas of high prevalence where the disease is less common among individuals of African or Asian descent.
The lifetime risk of developing MS in a high prevalence area is 1 in 400 among individuals of European descent. The mean age of onset is about 30 years and the disease is 2 - 3 times more frequent in women.
2. Classification and natural history
Pathologically, MS is a chronic, ongoing neurological disorder. Clinically, however, certain phases and landmarks are recognised (Table 1). With the increasing availability of MRI, patients undergoing scans for other reasons may incidentally be found to have subclinical lesions consistent with MS[3] and to fulfil the Barkhof-Tintore radiological criteria.[4,5] These patients are considered to have radiologically isolated syndrome (RIS); about one-third develop clinical signs within 2 years.[6,7]
When patients present for the first time with symptoms and signs of a possible demyelinating disorder, they are considered to have a clinically isolated syndrome (CIS). The presentation may be mono- or multifocal and the disease usually targets the optic nerve (25% of cases), brainstem (15%) or spinal cord (50%).[8] Prior to the advent of MRI, clinical studies estimated the risk of progression to clinically definite MS (CDMS) in high-risk regions, such as the United Kingdom (UK) and US, to range from 30% to 75%. MRI has allowed for an earlier estimation of risk, which may have therapeutic implications. In a 10-year follow-up of CIS patients with an abnormal MRI, over 80% converted to CDMS, while only 11% with a normal MRI progressed to CDMS.[8] Cerebrospinal fluid (CSF) and other paraclinical data may also predict which patients with CIS will go on to develop relapse remitting MS (RRMS). The presence of oligoclonal bands (OCBs) in the CSF had a positive predictive value of developing MS of 97%, a sensitivity of 91% and a specificity of 94%.[9] The hazard ratio (HR) for developing CDMS - if all 3 evoked potentials are abnormal - is 1.6 (95% confidence interval 0.8 - 3.2).[10] Almost 85% of MS patients have an RRMS course. A relapse refers to the development of symptoms lasting for at least 24 hours. All symptoms occurring within 1 month are regarded as constituting a single attack. If these episodes or attacks of neurological symptoms in the early stages of the disease reverse fully, then the patient is then said to be in remission. After an average of 10 - 12 years, most patients with RRMS will enter a progressive phase of permanent neurological disability with or without superimposed relapses.[11] This phase is referred to as secondary progressive MS (SPMS). About 15% of patients have progressive unrelenting disease from the onset. This category is referred to as primary progressive MS (PPMS).[12] Some of the risk factors and predictions for the development of the various stages and far greater disability are summarised in Fig. 1. Greater attack frequency has been linked to a low vitamin D level, non-white race and MRI lesion load. Non-whites are more likely to develop greater attack severity and poor attack recovery.[13,14] About half of all patients will have cognitive impairment. Depression is 3 times more common and the suicide risk is doubled. Life expectancy is reduced by 10 - 15 years and is usually due to secondary infections of the chest and bladder, and pressure sores.

2.1 Assessment of disability
The most widely-used scale for disabilities is the Kurtzke Expanded Disability Severity Scale (EDSS) (Table 2). While useful in monitoring therapy (see later), it provides little information about upper-limb function and gives no information about cognition. The more important landmark scores are 3 (considering most drug trials enter patients who have an EDSS <5.5), 6 (the need for an aid to ambulate), 8 (wheelchair- or bed-bound) and 10 (death).
As the EDSS focuses on ambulation only, other scales have been devised to assess parameters, such as upper-limb function and cognition. One such example is the MS Functional Composite (MSFC), the details of which can be downloaded from the National MS Society website (http://www.nationalmssociety.org). The MSFC tests ambulation by a timed 25-foot walk 'test', upper-limb function by the 9-holed peg test and cognition by the paced auditory serial addition test (PASAT).
3. Diagnosis
3.1 Introduction
There is no single symptom, sign or test that will confirm a diagnosis of MS. In earlier times, the diagnosis required 2 separate clinical episodes disseminated in time (DIT) at least 1 month apart and disseminated in space (DIS). There should be no other explanation for the constellation of symptoms and signs. Important presenting symptoms include: sensory disturbances such as numbness, paresthesia, band-like sensations; visual symptoms such as unilateral visual loss and diplopia; and disturbance of gait and sphincters. Over time, the paraclinical tests of imaging, CSF examination and evoked potentials have been included to make a more secure diagnosis of MS (diagnostic criteria).
3.1.1 CSF analysis
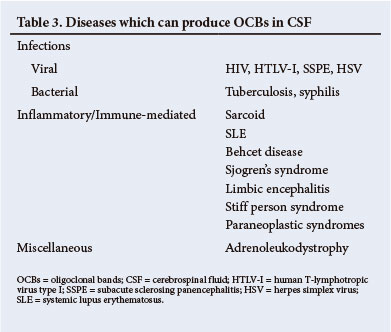
The most important abnormality in the CSF is the presence of intrathecal immunoglobulin synthesis, which may be detected either by the presence of OCBs or an increased immunoglobulin G (IgG) index. OCBs consist of proteins that are separated electrophoretically in paired CSF and serum samples. The best technique is isoelectric focusing. For a positive result, there must be >2 bands that occur exclusively in the CSF and not in the serum. Over 95% of patients with MS will have OCBs in the CSF.[15] This test is superior to the IgG index, which is abnormal in only 70% of patients.
It must be emphasised that a positive result must be interpreted in the context of the clinical diagnosis. A number of other disorders, notably infections, can also lead to the presence of OCBs in the CSF (Table 3). A negative OCBs result should alert one to review the diagnosis of MS.[15]
3.1.2 Evoked potentials
Evoked potentials are most useful in providing evidence of a clinically silent lesion. The usual ones tested are the visual evoked potentials (VEPs), brainstem auditory evoked potentials (BAEPs) and somatosensory evoked potentials (SSEPs). Of these, the VEPs are the most useful.
3.1.3 MRI
MRI represents a major advance in the diagnosis of MS. It allows for an earlier and more confident diagnosis and plays a major role in the diagnostic criteria for MS (see later). Certain shapes and distribution of white matter lesions (WMLs) have a high sensitivity and specificity.[14,51] The lesions tend to be ovoid, usually perpendicular to the lateral ventricles, maybe juxtacortical, corpus callosum, infratentorial or spinal. Enhancing lesions indicate active disease. While MRI has become an indispensable tool in the diagnosis of MS, it must be emphasised that there are many causes of WMLs (Table 4). Without careful attention to the clinical presentation and the MRI lesion pattern, MS may be wrongly diagnosed to the patient's detriment.

3.2 MRI examination of possible MS
3.2.1 Revised McDonald Diagnostic Criteria (2010) (Table 5)
• DIS: >1 T2 lesion in at least 2 of the 4 following areas of the central nervous system (CNS): periventricular, juxtacortical, infratentorial or spinal cord.
• DIT: there is a simultaneous presence of asymptomatic gadolinium (Gd)-enhancing and non-enhancing lesions at any time or a new T2 and/or Gd-enhancing lesion, or lesions on follow-up MRI which can be performed at any time.
3.2.2 Technique
3.2.2.1 Brain
• Transverse fluid attenuated inversion recovery (FLAIR)
• Coronal T2
• Sagittal T1 and sagittal T2
• Diffusion sensitive sequence
• Sagittal T2 of cervical and thoracic cord (if clinically indicated).
3.2.2.2 Post contrast
If any lesions are found in the typical locations (see the McDonald Criteria above), then post-contrast sagittal, coronal and transverse T1 images should be obtained. If any typical spinal cord lesions are demonstrated, then a pre-contrast T1 sequence, probably axial, should be obtained, followed by sagittal and axial post-contrast sequences.
3.2.2.3 Signal pattern of lesions
• T1: hypo- or iso-intense. Marked hypo-intensity correlates with chronicity.
• T2: mild to moderately hyper-intense.
3.2.2.4 Morphology and location
• Periventricular lesions. Lesions are 'applied' to the margins of ventricular structures, without intervening tissue. Lesions can be localised to any ventricular surface, but in particular to the lateral surface of the temporal horns, the trigones and the frontal horns.
• Periventricular lesions are also noted in relation to the corpus callosum, in particular the calloseptal interface. Lesions of the inferior surface of the central corpus callosum should also be searched for on the sagittal T2 sequence, as these are quite specific for MS.
• Juxtacortical lesions are located within the deeper layers of the cortex, and the interface between the cortex and underlying white matter.
• Infratentorial lesions are noted in particular in the middle cerebellar peduncles, but also in the deep cerebellar white matter and applied to the ventricular margins of the 4th ventricle.
• Spinal cord. The cervical spinal cord is most commonly affected. Lesions are noted in the dorsolateral aspect of the cord, encompassing less than half of the cross-sectional area of the cord, and <2 vertebral segments (usually only 1 vertebral segment).
• Post-contrast enhancement of lesions could be nodular, encompassing the majority of the lesion, a ring or semilunar, incorporating a portion of the circumference of the lesion. Similar enhancement patterns occur in spinal cord lesions.
3.2.2.5 Contrast dosage and timing
• The usual standard dose is 0.05 mmol/kg. Some institutions administer 0.1 mmol/kg. A triple dose (TD) would be 0.3 mmol/kg.
• Most authors advise a single dose to be administered. If an enhancing lesion was demonstrated, then the criteria regarding enhancement will have been met.
• Some contrast agents (e.g. gadobutrol) demonstrate progressive enhancement of MS lesions at 5 - 10 minutes post-injection; therefore, a delay of 10 minutes should occur between contrast administration and scanning.
• Other Gd-containing contrast agents demonstrate varying degrees of delayed progressive enhancement. Obtaining 3 separate post-contrast sequences would result in the last sequence being obtained at the time that delayed enhancement would be expected. The final post-contrast images should therefore be especially well scrutinised.
• In our opinion, a practical approach would be that, if enhancing lesions are still not noted, another standard dose should be administered, depending on clinical indication, e.g. first episode with a strong suspicion of MS.
• A TD is usually not advised. More lesions are noted following a TD, but no lesions are noted after only utilisation of a TD.
3.2.3 Diagnostic criteria
Over the years there have been several attempts to develop diagnostic criteria for MS since there is no single pathognomonic diagnostic test. The first important attempt was that by Schumacher et al.,[16] who used clinical criteria alone, including:
• age of onset of between 10 and 50 years
• objective clinical abnormalities
• symptoms and signs localised in the CNS
• DIS and DIT
• no other diagnosis.
In 1983, Poser et al.[17] introduced paraclinical findings to improve the reliability of the diagnosis. They added a category of laboratory supported MS if OCBs were present in the CSF, or the IgG index was raised. In 2001, McDonald et al.[18] used MRI in what became known as the McDonald Criteria. These criteria were modified in 2005[19] and again in 2010.[20] Further discussion will focus on these latest criteria (Table 5). Based on the findings of the MRI research group,[21-23] the 2010 panel refined the McDonald Criteria to allow an earlier diagnosis of MS when patients present with the first attack (CIS), using a single MRI study without losing specificity. These criteria are applicable only if the clinical presentation is typical and there is no better explanation for the patient's symptomatology.
Provided the above prerequisites are met, DIS and DIT can be shown on a single MRI scan. DIS can be demonstrated by >1 T2 lesion in at least 2 of the following 4 areas: periventricular, juxtacortical, infratentorial and spinal cord. If the subject has a symptomatic brainstem or spinal cord lesion, these are not counted. DIT can be shown on a single scan if there are simultaneous asymptomatic Gd-enhancing and non-enhancing lesions. Alternatively, DIT may be shown if a subsequent scan done at any time shows a new T2 and/or Gd-enhancing lesion(s). For PPMS there should be disease progression of at least 1 year and 2 of the following criteria:
• evidence of DIS in the brain at >1 T2 lesion in a typical site
• evidence of DIS in spinal cord >2 T2 lesions (symptomatic brainstem and spinal cord lesions are excluded)
• positive OCBs in the CSF or elevated IgG index.
4. Differential diagnosis
4.1 Introduction
Before making a diagnosis of MS it is imperative that alternative diagnoses are considered and excluded. This is particularly important in South Africa (SA), which is a moderate prevalence MS area, has a high burden of neurological infections and where the majority of the people are black - an ethnic group that has a very low frequency of MS. Furthermore, before applying the diagnostic criteria, there should be no better explanation for the patient's presentation.[20]
The differential diagnosis is very wide and the reader is referred to a detailed review.[24] The majority of the other diagnoses can easily be excluded by careful clinical assessment alone. Some of the more important and relevant diagnoses in the Southern African context are listed in Table 6. A few are discussed in more detail.

4.1.1 Acute disseminated encephalomyelitis
Acute disseminated encephalomyelitis (ADEM) is a monophasic illness, which is usually preceded by an infection. Viral exanthemas are the most frequent cause of ADEM. However, ADEM may follow other infections such as HIV, trauma and immunisation. The presentation is poly-symptomatic and consists of varying combinations of optic nerve, brainstem, spinal cord and cerebral hemisphere dysfunction. This initial event may be difficult to separate from CIS, but suggestive features include an encephalopathy, bilateral visual impairment, neuroretinitis or complete transverse myelopathy. The encephalopathy may manifest as disturbed level of consciousness, altered behaviour or cognitive deficits. The course may last from 1 week to 3 months with variable recovery. MRI may show lesions of varying size including large ones, situated supra- and infratentorial, even in the basal ganglia. Transient OCBs may occur. 'Relapses' of the identical clinical and MRI features are referred to as multiphasic disseminated encephalomyelitis (MDEM). When a 'relapse' occurs with new signs and new MRI lesions, the disease is referred to as recurrent ADEM, but this term is problematic and the patient may really have MS.[24]
4.1.2 Neuromyelitis optica (NMO)
The combination of the bilateral visual impairment and a myelopathy may be seen in a variety of clinical disorders such as HIV, syphilis, sarcoid and systemic lupus erythematosus (SLE). The idiopathic variety, previously often referred to as opticospinal MS, has now been shown to be a distinct disorder associated with anti-aquaporin 4 antibodies.[25]
The original description of idiopathic NMO was that of a monophasic illness characterised by severe bilateral visual loss and a severe myelopathy. Following the discovery of the antibody, it has become clear that patients may have recurrent episodes, blurring the distinction from RRMS. The spectrum of clinical features has widened to include recurrent optic neuritis alone, recurrent myelitis alone, intractable vomiting, intractable hiccups and other brain presentations. MRI features suggestive of antibody-positive NMO include a longitudinal myelitis spanning at least 3 vertebral segments and brain lesions adjacent to the ventricular system, or the hypothalamus and dorsal medulla. Other WMLs atypical for MS may be present. OCBs are detected in 10 - 20% of patients.
The revised Wingerchuk Criteria[26] require the presence of optic neuritis, myelitis and 2 of 3 paraclinical tests, namely: contiguous spinal lesion of at least 3 vertebral segments in length, non-specific brain changes at onset and anti-aquaporin 4 antibody positivity. The distinction from MS is important as NMO responds to immunosuppressive therapy and not to the immunomodulatory drugs used in MS.
4.1.3 Differential diagnosis in the SA patient
For reasons outlined earlier, apart from identifying clinical and paraclinical features that suggest a diagnosis of MS, parallel investigations have to be undertaken to exclude diagnoses that may mimic MS. The minimum investigations that should be undertaken in a suspected case of MS are outlined in Table 7.
5. MS treatment target group
Treatment of MS has become possible with the development of current disease-modifying therapies (DMTs), which have a major influence on those living with this disease.[27]
It is important to understand that patients with MS more often than not, will continue to experience disease activity despite treatment, and current DMT only alters the natural history (progression) of this neurodegenerative disorder, and is not curative.
The aim of identifying a target group of patients with a diagnosis of MS would be to clearly define a subgroup of patients with a definite diagnosis of MS who will benefit from the initiation or continued use of DMT, and these guidelines will be supported by current scientific evidence.
This will enable the prescribing specialists (neurologists and specialist physicians) to justify initiation and continuation of treatment for this defined 'target group' of MS patients. The target group will be defined by certain entry and exit criteria - agreed upon by the SA MS Advisory Board - and all prescribing doctors will be advised to adhere strictly to these proposed guidelines of treatment.
The entry and exit criteria would target those patients who would potentially benefit most from DMT, and to exclude those patients for whom there is no scientifically proven benefit from either initiating or continuing expensive DMT.[37]
The renewed diagnostic criteria for MS (McDonald Criteria, 2010)[20] substantially modified the concept of a CIS, since the diagnosis can now be made at first presentation in 30 - 50% of patients, and cases of true CIS would therefore be limited.[20]
Previous studies assessing the benefit of initiating DMT in patients with high-risk CIS, did this based on previous MS criteria (McDonald Criteria, 2005); therefore, it is important to understand that most of these patients would these days be classified as MRI-definite MS and no longer as CIS (ETOMS[30]/REFLEX[31]).
Based on current knowledge, we propose the following entry and exit criteria.
5.1 Criteria for initiating and continuing therapy
5.1.1 Inclusion criteria
• Definite RRMS based on revised McDonald Criteria
• High-risk CIS
• Diagnosed patients with relapsing SPMS and EDSS of <6.5
• If a patient with advanced non-relapsing SPMS should present with clinical evidence of a relapse in combination with MRI evidence of a contrast-enhancing lesion (relapse) after discontinuation of treatment (DMT should be reinitiated).
5.1.2 Non-initiation criteria
• PPMS
• Low-risk CIS
• EDSS >6 (non-relapsing SPMS) sustained for a period of 12 months
5.2 Exit criteria
It is very difficult to ascertain whether a patient is no longer benefitting from their treatment and the problem of discontinuation is made more problematic as few alternatives exist for disease modification. Thus, mandatory stopping criteria that apply to all cases are not feasible. The following are suggestive of loss of, or limited benefit from, treatment and should be taken into account when deciding whether treatment should be discontinued:
1. Continued relapses are indicative of suboptimal disease control; treatment should be switched or escalated and not discontinued. Only where relapses continue to occur at similar pre-treatment frequencies, despite the use of all the higher-tier treatment options, should discontinuation be considered.
2. Therapy should be discontinued (not switched or escalated) when individuals are unlikely to benefit because of their more advanced disability level (i.e. non-ambulatory, EDSS >6.5 for 1 year). However, disproportionate spinal cord involvement occurs in a small percentage of patients. It is thus the Advisory Board's opinion that a patient who still has relapsing disease, good cognition and good upper limb dexterity and function, may benefit from treatment despite loss of ambulation.
3. The benefit of current therapies can potentially be maximised during the predominantly early inflammatory phase, from which MS gradually evolves into a more degenerative phase. Where no further relapses occur and the patient continues to experience disease progression (defined by an increase of 2 EDSS points), discontinuation should be considered. However, should relapses then recur, treatment should be restarted.[28]
It is important to monitor patients clinically and radiologically on a 12-monthly basis to ensure that they continue to qualify for a DMT - considering the exit criteria.
5.3 Considerations 5.3.1 CIS
We propose to delay starting DMT in lower-risk patients until the disease is obviously active (either follow-up MRI or clinical progression to definite MS, revised McDonald Criteria, 2010).[29]
There may be a concern regarding the possibility of disease activity despite a lack of clinical and MRI findings - but data from the 5-year extension studies of intramuscular interferon (IFN)-beta-1a (30 µg) once a week or subcutaneous (SC) IFN-beta-1b (250 µg) every other day in patients with CIS did not support this concern. The delay in starting IFN-beta by a median of 29 months or 23 months did not result in a lower rate of confirmed disability progression in the early treatment groups.[31,32]
5.3.2 High-risk CIS
High-risk CIS is defined in patients who present with clinical and MRI features highly suggestive of MS, but do not fulfil the criteria of the revised McDonald Criteria to qualify as having MRI or CDMS. However, these patients will have a positive VEP or positive CSF markers.
In patients with true 'high-risk CIS', one could consider a different approach, based on the results of the REFLEX study, and perhaps initiate a cost-effective alternative to standard MS treatment, and opt for SC IFN-beta-1a (44 µg) once a week.
5.3.3 SPMS over a period of 6 - 12 months
Many patients develop SPMS with significant disability after 10 - 15 years of illness and continue to use DMTs when they are no longer clinically effective.[34,35]
The UK MS guidelines indicate the need to stop first-line DMTs in a patient with SPMS with the development of the loss of the ability to walk with or without aids and persisting over a period of 12 months.[36]
Guidelines from the US conclude that no evidence of benefit from IFN-beta was apparent in patients with sustained EDSS of 6.5.[37]
It is important to introduce the term of relapsing and non-relapsing SPMS to decide on future management.
The Association of British Neurologists (ABN) Guidelines acknowledge that it can be very difficult in some cases to conclude that 'treatment is providing (absolutely) no benefit and the problem of discontinuation is compounded by' the lack of alternative therapeutic options for disease modification in patients with disease progression and disability.[36]
The ABN guidelines conclude that it may not be 'feasible to have mandatory stopping criteria that apply in all cases'.[36] One approach to this problem would be to indicate clearly to patients at the time of initiation of treatment that they are being prescribed DMT for RRMS and that once they have entered the SPMS phase without relapses and have an EDSS level of >6.5 for 1 year, DMT must be stopped.
5.3.4 PPMS
DMTs are used to suppress inflammatory relapses and have never been shown to be therapeutic in PPMS to date.[38]
The MS Advisory Board will review the definition of the target group on an annual basis.
6. Treatment
The more common drugs for treatment of MS are listed in Table 8.
6.1 Managing the acute relapse
The acute attack is the main reason patients present initially and subsequently to the treating doctor.
Glucocorticoids have remained the mainstay of treatment for the acute attack as they have potent anti-inflammatory properties and restore the integrity of the blood-brain barrier (BBB). They have been shown in many trials to be more effective than placebo. They induce T-cell apoptosis and diminish proinflammatory cytokines. Only one multicentre placebo-controlled trial has been done to show the efficacy of steroids in treatment of the acute relapse in MS.[39]
Intravenous methyl prednisolone (IVMP) has been shown to be more effective than orally administered IVMP based on the optic neuritis treatment trial that showed that 1 g of IVMP was most effective. There are no data to indicate that steroids prevent further attack or alter long-term disability.
Current dosing is 1 g of IVMP for 3 days or 500 mg IVMP for 5 days. The need for subsequent oral tapering therapy is unnecessary and depends on the treating physician. The American Academy of Neurology (AAN) classifies the treatment of an acute attack of MS with glucocorticoids as a type A recommendation.
For steroid non-responders or reoccurring relapses/progression despite treatment with high-dose steroids, there are 2 other treatment options. In a recent survey of non-responders, 41% of patients responded to plasma exchange with marked functional improvement; 400 mg/kg/day hyperimmune globulin for 5 days has also been tried with favourable outcome.
Both options have a category II indication as supportive or adjunctive treatment to glucocorticoids.
Mitoxantrone can be administered as an induction agent in fulminant aggressive MS attacks (see mitoxantrone therapy, section 6.3.1).
6.2 First-tier DMT
Relapsing MS is amenable to treatment and should be initiated as soon as this form of MS has been confirmed. The revised McDonald Criteria (2010) aimed to identify these individuals with high specificity.
IFN-beta-1a (Rebif and Avonex), IFN-beta-1b (Betaferon) and glatiramer acetate (GA) (Copaxone) are indicated as first-tier treatments (Fig. 2). These have been demonstrated to have moderate efficacy, but excellent safety.
6.2.1 Mechanism of action
IFN-beta is produced by fibroblasts in vivo. It has antiviral, antiproliferative and immunomodulatory effects. IFN-beta-1b differs slightly from the natural IFN-beta and is not glycosylated, whereas IFN-beta-1a represents the natural amino acid sequence and is glycosylated. IFN-beta-1a and IFN-beta-1b modulate the immune system through a number of mechanisms:[40]
1. They down-regulate the level of expression of major histo-compatibility complex (MHC) class II molecules as well as the cluster of differentiation (CD) 20 and CD28 co-stimulatory molecules on antigen-presenting cells (APCs). This results in reduced T-cell activation.
2. IFN-beta reduces the expression of T-helper (TH) 1 pro-inflammatory cytokines (specifically IL-2, IL-12 and IFN-gamma) and increases the expression of TH2 stimulatory cytokines (IL-4 and IL-10). The immune response is consequently shifted towards a TH2 profile, thus away from the cell-mediated immune response characteristic of MS.
3. Activated T-cells express very late antigen (VLA)-4, which binds endothelial vascular cell adhesion molecule (VCAM)-1. This binding results in T-cell secretion of matrix metalloproteinases (MMPs), which degrade the extracellular matrix and allow T-cell migration through the BBB. IFN-beta has been demonstrated to reduce expression of VLA-4, reduce the secretion of MMPs and causes shedding of soluble VCAM-1. 4. IFN-beta appears to restore suppressor cell function.
GA is an oligopeptide consisting of the amino acids L-tyrosine, L-glutamine, L-alanine and L-lysine.[41] The oligopeptide length ranges from 40 to 100 residues and the sequence is random. GA was synthesised to resemble myelin basic protein (MBP), which was a suspected auto-antigen in MS. However, it was found that in murine experiments where it was expected to induce experimental autoimmune encephalitis, it had a protective effect. Subsequently it has been developed as a therapeutic drug. The mechanism of action is still uncertain.
1. GA has been demonstrated to induce populations of TH2 cells reactive to GA. These cells then enter the CNS and cross-react with myelin. This appears to result in the release of anti-inflammatory cytokines.
2. Regulatory T-cells are also activated and GA-reactive CD8+ cell levels are restored; these are both significantly reduced in the untreated MS population.
3. GA suppresses the innate immune system directly by alteration of APC function.
4. GA results in the differentiation of CNS APCs, such as microglia, into type II APCs. These then favour TH2 deviation.
6.2.2 Summary of clinical trials
The trials summarised in the following sections support these conclusions:
1. The 4 available first-tier agents are beneficial in RRMS.
2. All of these treatments significantly delay the onset of CDMS in subjects with CIS.
3. IFN-beta-1a (Rebif) and IFN-beta-1b (Betaferon) are beneficial in SPMS, specifically where superimposed relapses occur.
4. The first-tier treatments are approximately similar in efficacy. Higher than the standard doses of IFN-beta confer no additional benefit.
5. Early initiation of treatment has long-term disease-modifying effect and results in reduced mortality.
6. Switching between treatments is beneficial in many cases and a viable option in the case of treatment failure.
6.2.3 RRMS
The following pivotal trials have clearly demonstrated efficacy in the RRMS population:
Betaferon was the first therapy to achieve US Food and Drug Administration (FDA) approval based on a pivotal trial (1993).[42] A total of 372 subjects were randomised to receive either placebo, low-dose (1.8 mU) or high-dose (8 mU) Betaferon on alternate days. At 2 years the annualised relapse rate (ARR) had improved in the high-dose arm (0.84) and in the low-dose arm (1.17), compared with placebo (1.27). There was also a significant difference between the high-dose and low-dose arms.
This trial was extended to 5 years to find that there was a lower proportion of patients with a sustained increase in disability in the 8 mU group (35%), compared with the placebo group (46%).
Avonex was demonstrated to be of benefit in another pivotal trial (1996).[43] A total of 301 subjects were randomised to placebo or Avonex - 30 µg intramuscular injection (IMI) weekly. At 2 years, the ARR was lower in the treatment arm (0.66) compared with the placebo arm (0.9). Fewer subjects on the treatment arm experienced disability progression (21.9%) compared with the placebo arm (34.9%).
Rebif's efficacy was demonstrated in the PRISMS trial (1998);[44]
560 subjects were randomised to placebo and received either 22 µg or 44 µg of Rebif 3 times per week (t.i.w.). At 2 years the ARR in the low-dose arm (22 µg Rebif) was 1.82, and the high-dose arm (44 µg Rebif) was 1.73, compared with placebo (2.56). Subjects on the treatment arms also experienced a delay in the progression of sustained disability - 21.3 months in the high-dose arm, 18.5 months in the low-dose arm and 11.9 months in the placebo arm. This trial was extended for a further 2 years, where the placebo arm was randomised to either the low or high dose. During this period the subjects who had been on the high-dose arm from the trial onset experienced a delay in disability progression of 18 months, compared with placebo.
GA (Copaxone) was found to be effective in a trial published in 1995.[45] A total of 251 subjects were randomised to receive either daily doses of 20 mg GA (SC) or placebo. At 2 years, the active arm experienced a reduction in ARR (0.59) compared with the placebo arm (0.84). Significantly more patients receiving GA improved and more receiving placebo worsened (p=0.037) when comparing the proportion of patients who had improved, were unchanged, had remained unchanged or had worsened by >1 EDSS from baseline to conclusion.
6.2.4 Adverse effects and neutralising antibodies
The most common side-effects of IFN-beta-1a and IFN-beta-1b are flu-like symptoms after injection and injection-site reactions. Hepatic transaminase enzyme elevation and leukopenia may occur; routine full blood count and liver function testing every 6 months is recommended. Injection site reactions are less common with the IMI of IFN-beta-1a (Avonex).
Neutralising antibodies may develop with use of the IFNs. This has been demonstrated to influence the efficacy of these treatments. However, the expert consensus report by the AAN in 2007 concluded that there are insufficient data to provide guidelines on neutralising antibody testing.
6.2.5 CIS
Following the initial pivotal trials, these drugs have been explored in patients with an isolated clinical demyelinating event or CIS.
CIS may occur with no radiological evidence of demyelination (e.g. optic neuritis), or with a single demyelinating lesion. DMT is inappropriate in this clinical scenario, since the differential diagnosis is wide and there is risk of recurrence and thus evolution to MS. CIS with radiological evidence of DIS and DIT, specifically the presence of non-contrast enhancing and asymptomatic contrast-enhancing lesions, now fulfil the revised McDonald Criteria (2010).[20]
The population selected for the CIS trials comprised subjects with an isolated clinical event and at least 2 asymptomatic lesions, thus CIS with DIS, and CIS with DIS and DIT.[20]
Avonex was evaluated in CIS in the CHAMPS trial (2000);[46] 383 subjects were randomised to receive either 30 µg Avonex IMI weekly or placebo. At 3 years there was a significant decrease in the risk of a second attack in the active arm (35%), compared with placebo (50%).
Rebif was tested in the ETOMS trial (2001);[30] 308 subjects were randomised to receive either 22 µg Rebif or placebo SC weekly. At 2 years, the risk of a further event was 34% in the active arm, compared with 45% in the placebo arm.
In January 2012, the new formulation Rebif was evaluated in the REFLEX trial;[31] 517 subjects with CIS were randomised into a high-dose arm (44 µg Rebif t.iw.), a low-dose arm (22 µg Rebif weekly) and a placebo arm. At 2 years, 62% of patients in the high-dose arm reached the McDonald Criteria (2005) for MS, compared with 76% in the low-dose arm and 86% in the placebo arm. The secondary endpoint was conversion to CDMS (Poser Diagnostic Criteria, 1983) implying a second attack - 21% in the high-dose arm, 22% in the low-dose arm and 38% in the placebo arm reached this endpoint.
Betaferon was evaluated in the BENEFIT trial (2006);[47] 292 subjects with CIS were randomised to receive either 8 MIU Betaferon or placebo on alternate days. At 2 years 26% of the active arm had suffered a second event, compared with 44% in the placebo arm.
GA was evaluated for CIS in the PRECISE trial (2009);[48] 481 subjects were randomised to receive either 20 mg of GA or placebo. At 2 years, 25% of the treatment arm had experienced a second attack compared with 43% in the placebo arm.
6.2.6 SPMS
Two first-line formulations have demonstrated efficacy in SPMS.
The European study group on IFN-beta-1b in SPMS published the following findings in 1998.[49] A total of 718 patients were randomised to receive either Betaferon or placebo. In the active arm progression was delayed by 9 - 12 months in the study period of 2 - 3 years.
The benefit was seen both in the group with superimposed relapses and also the group with only progressive disease.
Rebif was investigated for this indication in the SPECTRIMS trial (2001),[50] 618 patients were randomised to receive either placebo, 22 µg Rebif t.i.w. or 44 µg Rebif t.i.w. over 3 years. Time to confirmed disability progression was not significantly improved in the active treatment arms. However, in subjects with relapses in the 2 years before inclusion in the trial, the delay in disability progression was more pronounced in the combined treatment arms (HR 0.74, p=0.055). There was a significant reduction in relapse rate in both active treatment arms (0.5) and in placebo (0.71).
6.2.7 Comparison of first-tier therapies and dosing regimes in RRMS
A number of trials have compared different first-tier therapies:
In the REGARD trial (2008),[51] 764 subjects were randomised to receive either GA or Rebif. At 96 weeks there was no difference in the time to first relapse, relapse rate or disability progression. However, there were significant differences in certain MRI parameters. Although there was no difference in the number of new or enlarging T2 lesions, the mean number of Gd-enhancing lesions per patient per scan was significantly less in the Rebif arm over the 96-week period. The number of new or enlarging T2 lesions and the number of contrast-enhancing lesions plateaued at 24 weeks in the Rebif arm, compared with 72 weeks in the GA arm. This is suggestive of a slower onset of action of GA.
In the BEYOND trial (2009),[52] 2 244 subjects were randomised to receive either high-dose Betaferon (16 MIU on alternate days), standard dose (8 MIU on alternate days) or GA 20 mg daily, in a 2:2:1 ratio. Subjects participated for at least 2 years, though the first subjects continued up to 3.5 years. There were no differences in relapse rate and EDSS progression. The increase in T2 lesion volume, on the last available MRI compared with screening, was significantly greater in both the Betaferon arms compared with the GA arm. This difference was more pronounced in the first year of treatment.
In the single blind INCOMIN trial (2002),[53]188 subjects were randomised to receive either 8 MIU Betaferon or 30 µg Avonex weekly. At 2 years, 51% in the Betaferon arm remained relapse free compared with 36% in the Avonex arm. There were also relatively fewer new T2 lesions and delays in disease progression in the Betaferon arm: 13% in the Betaferon arm experienced a 1-point progression in EDSS sustained for 6 months, compared with 30% in the Avonex arm. Later trials, though smaller or of a shorter duration, were not able to reproduce this result.
In the single blind EVIDENCE trial (2002),[54] 677 subjects were randomised to receive either 44 µg Rebif t.i.w. or 30 µg Avonex weekly. The Rebif arm experienced fewer relapses and T2 lesions at 48 weeks, with the difference maximal in the first 24 weeks. There was no difference in disability progression. Neutralising antibodies were detected in 25% of patients in the Rebif arm and 2% in the Avonex arm.
No benefit in increased dose of either IFN-beta-1a or IFN-beta-1b has been found in the following trials:
The BEYOND trial[52] revealed no difference in relapse rate, EDSS progression or MRI parameters between the 8 MIU and 16 MIU alternate day Betaferon arms.
In 802 subjects, randomised to receive either 30 µg or 60 µg Avonex weekly for at least 3 years, there was no difference in clinical or MRI measures.[51]
6.2.8 Long-term efficacy of first-tier therapies
The following trials illustrate the long-term beneficial effect of early treatment.
The CHAMPS trial for CIS (2000), was extended to 5 years and 10 years; 81 subjects in the active arm and 74 subjects in the placebo arm were included in the extension trial, CHAMPIONS.[56]
All patients were treated with 30 µg Avonex weekly. Treatment was delayed in the placebo arm (thus delayed treatment arm) by a mean of 30 months. At 10 years, the risk of CDMS was lower (58% v. 69%, respectively) and the ARR was lower (0.16 v. 0.33, respectively) in the immediate treatment arm compared with the delayed treatment arm. There was no difference in disability. At 10 years, 9% had reached an EDSS of >4, and 6% an EDSS of >6.
Subjects who participated in the initial pivotal IFN-beta-1b trial, which extended to 5 years, were followed up for a median of 21.1 years after initial enrolment.[57] Treatment after the initial trial period was dependent on the discretion of the treating physician. The mortality rate was 18% (22/122) in the high-dose arm, 17.9% (22/123) in the low-dose arm and 30.6% (37/121) in the placebo arm. The HR of death by Kaplan-Meier estimates was reduced by 46.8% in the high-dose arm compared with the placebo group.
6.2.9 Switching between first-tier therapies
Switching between therapies should be considered when the current treatment has proven to be ineffective, or when the adverse effects are significant.
Switching from GA to an IFN-beta or vice versa and switching from one IFN-beta to another appears to be beneficial, demonstrated by the following trials:
• Coan et al.[58] reported the clinical course of 85 subjects with RRMS who had received Avonex for at least 18 months and opted to switch to GA. These subjects were followed for 36 - 42 months. Sixty-two switched due to lack of efficacy and experienced a reduction of ARR from 1.32 to 0.52. A further 23 switched due to side-effects and experienced a reduction of ARR from 0.62 to 0.47.
• Gajofatto et al.[59] reported 101 subjects who switched between first-tier agents due to lack of response. At 2 years the subjects who changed from an IFN-beta to GA had ARR reduced from 0.55 to 0.25. In subjects who changed from GA to IFN-beta the ARR reduced from 0.5 to 0 and from one IFN-beta to another the ARR reduced from 0.68 to 0. The authors concluded that 'switching first-line DMT in patients with RRMS failing initial therapy may be effective in many cases. At least 2 other trials revealed similar results'.[60,61]
6.2.10 Recommendations
The MS Advisory Board recommends the following regarding the choice of first-tier DMT:
1. Treatment with a DMT should be initiated as soon as the diagnosis of RRMS is confirmed with the revised McDonald Criteria (2010).
2. Initiation with either IFN-beta-1a or IFN-beta-1b. GA should preferably be reserved as switch therapy. This recommendation is based on the delayed radiological disease modification observed with GA compared with IFNs[51,52] and the less frequent dosing schedule of the IFNs.
3. Where relapses continue to occur, switching between the first-tier treatments should be considered. The following switches are sensible: from an IFN to GA, from Betaferon to Rebif or vice versa and from low-dose IFN (Avonex) to either Betaferon or Rebif. If disease control is not obtained after switching, escalation to second-tier therapy is essential. The patient must not be switched more than twice before escalation to second tier DMT.
4. Switching can also be considered in cases of poor tolerance.
5. It is essential that the patient is not switched if the disease is well controlled.
6.3 Second-tier DMT
The presence of relapses signifies active inflammatory disease and necessitates either the introduction or change of DMT. If relapses continue despite adequate first-tier treatment, escalation of therapy is indicated.
The definition of a non-responder remains controversial. Often patients continue to suffer mild relapses with good recovery, but with a reduced frequency compared with the period before initiation of therapy. These patients do respond to treatment, but are not disease free. Freedom of disease remains the ultimate goal.
The European Medicines Agency (EMA) has adopted the following criteria for escalation of therapy, either to fingolimod or natalizumab (Fig. 2):[62,63]
'Adult patients aged >18 years with high disease activity despite treatment with beta-IFN. These patients may be defined as those who have failed to respond to a full and adequate course (normally at least 1 year of treatment) of beta-IFN. Patients should have had at least 1 relapse in the previous year while on therapy, and have at least 9 T2-hyperintense lesions in cranial MRI or at least 1 Gd-enhancing lesion. A "non-responder" could also be defined as a patient with an unchanged or increased relapse rate or with ongoing severe relapses, as compared with the previous year.'
This is also the definition submitted to the SA Medicines Control Council (MCC) for natalizumab. Currently in SA, a number of immunosuppressive treatments are being used in non-responders, including mitoxantrone, cyclophosphamide, azathioprine, methotrexate and regular pulses of methylprednisolone. Of these, only mitoxantrone has at least class II evidence of efficacy, according to the Therapeutics and Technology Assessment Subcommittee (TTAS) of the AAN.[64] The other therapies are not recommended for routine use in MS by this Advisory Board.
Currently, natalizumab has received approval and fingolimod is pending approval by the SA MCC. The following therapies have proven efficacy.
6.3.1 Mitoxantrone
Mitoxantrone is a synthetic antineoplastic anthracenedione initially approved for the treatment of acute myeloid leukaemia in 1987. It is a small molecule that crosses the BBB where it inhibits DNA replication and DNA-dependent RNA synthesis. It also inhibits the ability of topoisomerase II to repair DNA. It has a broad immunosuppressive effect, targeting predominantly the proliferating immune cells.
The MIMS trial was a double-blind placebo-controlled trial published in 2002;[65] a total of 194 patients with SPMS or PPMS were randomised to receive either placebo, 5 mg/m2 or 12 mg/m2 mitoxantrone, every 3 months for 2 years. The 12 mg/m2 group experienced a reduction of disability progression of 64% and reduction in ARR of 60%, compared with placebo.
In an Italian study (1997), 51 patients with RRMS were randomised to receive either 8 mg/m2 mitoxantrone or placebo every month for 1 year.[66] The active arm experienced a 79% reduction in disability progression of EDSS (2/27 active patients v. 9/24 of placebo patients) and 60% reduction in relapse rate.
A French\British trial (1997)[67] consisted of 42 patients with very active RRMS or SPMS randomised to receive either 1 g of methylprednisolone monthly or 1 g methylprednisolone with 20 mg of mitoxantrone monthly, for 6 months. The mitoxantrone arm experienced a reduction in the proportion of patients with enhancing lesions of 86%, and also a reduction in disability progression of 84% and relapse rate of 77%.
Mitoxantrone has also been demonstrated to be effective as induction therapy in patients with highly-active MS in the following trials.
In an observational study of 50 patients naive to DMT,[68] it has been demonstrated that monthly mitoxantrone infusions for 6 months, followed by IFN-beta had a sustained clinical benefit of up to 5 years, compared with IFN-beta alone. A similar result was obtained with mitoxantrone, followed by GA.[69]
In December 2011, Edan et al.[70] published a trial where 109 active, relapsing MS patients were randomised to receive either 12 mg/m2 mitoxantrone and 1 g methylprednisolone monthly for 6 months followed by IFN-beta-1b for the last 27 months, or methylprednisolone alone with IFN-beta-1b for the entire 36 months. The following results were reported: the 3-year risk of worsening disability was reduced by 65% in the mitoxantrone group relative to the IFN group (11.8% v. 33.6%, respectively). The mitoxantrone group had a reduced relapse rate (61.7%), a reduced number of Gd-enhancing lesions at month 9 and a slower accumulation of new T2 lesions at each time-point.
6.3.1.1 Risks of mitoxantrone
Potential cardiotoxicity was the first recognised significant risk associated with mitoxantrone treatment. The first sign of cardiotoxicity is a reduction of the left ventricular ejection fraction. Significant cardiotoxicity occurs at higher cumulative doses (thus >96 - 140 mg/m2). Clinical heart failure occurs in 0.4% of patients, as determined by the TTAS of the AAN.[68] However, subclinical impairment of the ejection fraction occurs more frequently (12% of patients) and also at lower cumulative doses (according to the TTAS). The FDA recommends that the ejection fraction is determined at baseline and before each treatment cycle.
If the ejection fraction decreases by 10% between evaluations or decreases to <50%, then the treatment must be discontinued.
Treatment-related acute leukaemia (TRAL) is an important potential complication of mitoxantrone treatment.[71,72] The risk was initially considered small, but it was later found to be between 1% and 12% where mitoxantrone was used for oncological indications. In 2000, the TTAS reported the incidence of TRAL to be 0.81% (37/4 076), based on the combination of reports where the denominator was available. TRAL occurs within the first few years after exposure, the longest reported delay is 5 years. An Italian group reports an incidence of 6.7/1 000.[71] The interval from onset of treatment to the development of TRAL was 3 years on average, with mean of 1.3 years from discontinuation of treatment (according to the Italian article). Haematological follow-up is thus essential for several years after treatment discontinuation.
Other significant adverse effects include nausea and vomiting, alopecia, increased risk of infections, amenorrhea, persistent amenorrhea (8%), infertility, modest increase in liver enzymes and bilirubin. Rarely, thrombocytopenia may occur.
6.3.2 Natalizumab
Natalizumab is an alpha-4-integrin antagonist. Binding of natalizumab to the alpha-4 beta-1 integrin on lymphocytes prevents binding to VCAM-1, and thus prevents migration of the lymphocyte across the BBB.
In the AFFIRM pivotal trial (2006),[73] 942 patients with RRMS were randomised in the ratio 2:1 to receive either 300 mg natalizumab by intravitreal injection every 4 weeks or placebo for 2 years. After 1 year, the ARR was 0.26 in the treatment group compared with 0.81 in the placebo group (68% relative reduction). This was maintained at the end of 2 years. At 2 years, the risk of probability of progression was 17% in the treatment group compared with 29% in the placebo group (42% relative reduction).
In the SENTINEL trial (2006),[74] 1 171 patients with RRMS who suffered at least 1 relapse in the past year on Avonex, were randomised to receive either addition of natalizumab to the Avonex arm, or placebo. At 1 year, the relapse rate in the combination group was 0.38 compared with 0.82 in the Avonex group (54% relative reduction). This was maintained at 2 years. EDSS progression occurred in 23% of the combination group and 29% of the treatment group.
In the GLANCE phase 2 trial (2009),[75] 110 patients who had received GA for at least 1 year and had suffered at least 1 relapse in that time were randomised to either the addition of natalizumab or placebo. The duration of the trial was 6 months, with 6 treatments of natalizumab. A 74% reduction in the number of Gd-enhancing lesions was found in the treatment group as well as 61% reduction in new or newly-enlarging T2 lesions. There was no significant difference in adverse events.
6.3.2.1 Risks of natalizumab[76]
Natalizumab was approved for the treatment of RRMS in 2004, but after 3 cases of progressive multifocal leukoencephalopathy (PML) were reported, the approval was suspended in February 2005. After further surveillance failed to reveal any additional cases of PML, the drug was approved as a monotherapy.
The TOUCH and TYGRIS programmes were initiated on reintroduction of natalizumab to monitor the incidence of PML.
By June 2011, 133 cases had been reported. All of these occurred after at least 1 year of treatment. The mortality rate was 18%. The overall risk is estimated to be 1.51/1 000 patients. The risk is 3 - 4 times higher in patients who have had previous exposure to immunosuppressive treatment at any time. The type and duration of treatment posing the greatest risk has not been established. In February 2011, the risk of PML in patients who had not used an immunosuppressant previously was 0.19/1 000 over the first 24 months and 1.37/1 000 after 24 months. In patients with prior immunosuppressant use the risk was 0.66/1 000 over the first 24 months and 4.3/1 000 thereafter.
Analysis of archived samples revealed that all patients who developed PML had positive serology for John Cunningham virus (JCV) before PML diagnosis (33 in one study[79] and 10 in another[80]). The risk of PML in patients with negative serology is thus presumed to be very low.
Natalizumab is, therefore, highly efficacious but with significant risk of developing PML, depending on previous immunosuppressive use and JCV serology. It is thus imperative that the drug is administrated by a physician familiar with the required long-term surveillance. A comprehensive surveillance and management protocol has been published. The following is a summary of this protocol:[78]
1. Before initiation of natalizumab treatment, a thorough baseline neurological examination and high-quality MRI must be performed. MRI must be repeated at least annually and more frequent MRI has also been suggested.
2. In patients receiving an immunosuppressive therapy, a 3 -6-month washout period has been proposed. A longer period is recommended for mitoxantrone and cyclophosphamide.
3. New neurological symptoms and signs should carefully be assessed. If not characteristic of MS, or if there is any doubt, an MRI must be performed.
4. If the clinical features and MRI features are compatible with a relapse, then the patient should be treated for a relapse. If the clinical status then stabilised or improves, then the patient can be routinely followed up.
5. If the clinical appearance or MRI suggests PML, or if the patient continues to progress despite treatment for a relapse, the diagnosis of PML should be considered. Natalizumab must then be suspended.
6. The diagnosis should be confirmed with CSF JCV polymerase chain reaction (PCR). However, if negative, the CSF should be repeated and other diagnoses must be excluded.
7. If the diagnosis of PML is confirmed, plasma exchange should be performed.
8. Patients with PML where natalizumab has been discontinued, and especially where plasma exchange has been performed, are at high risk of immune reconstitution inflammatory syndrome (IRIS). This occurs from 8 days to 12 weeks after plasma exchange. It manifests as marked neurological deterioration with Gd enhancement on MRI. These patients have generally been treated with high-dose methylprednisolone.
Natalizumab must be administered in a hospital or infusion centre. Patients experience mild infusion reactions including headache, dizziness and nausea, which respond to paracetamol, loratadine and slowing of the infusion rate. Hypersensitivity or allergic reactions rarely occur. In the AFFIRM trial,[75] these were also defined as anaphylactic or anaphylactoid reactions, urticaria, allergic dermatitis or hives. The hypersensitivity reactions occurred in 4% of patients in the AFFIRM trial. The current recommendation is to discontinue the infusion and not retreat the patient; however, this recommendation is being reviewed and pre-treatment with hydrocortisone and dexchlorpheniramine appears to prevent these reactions.
Interruption of treatment with natalizumab results in recurrence of MS disease activity. The degree of disease activity has not been demonstrated to be more than before initiation of therapy.[79] Disease activity increases shortly after interruption and peaks at 4 - 7 months.
6.3.3 Fingolimod
Sphingosine-1-phosphate (S1P) acts as a chemo-attractant for naive and central memory T-lymphocytes. These lymphocytes are drawn from lymph nodes by a relatively higher concentration of S1P in the circulation than in the interior of lymph nodes. Fingolimod binds to the S1P receptor on T-lymphocytes. Binding of fingolimod eventually results in internalisation of the receptor. These lymphocytes are consequently insensitive to circulating S1P and thus remain within lymph nodes. Fingolimod results in a reduction in the total mean lymphocyte count, but spares circulating effector memory T-cells. These cells lack chemokine receptor (CCR) 7, which is responsible for migration to lymph nodes. The effector memory T-cells may down-regulate the autoimmune response.
There are 5 S1P receptors, located on lymphocytes, neurons, oligodendrocytes, astrocytes and microglial cells. Fingolimod binds to 4 of the 5 receptors. It is thought that fingolimod may have a beneficial modulatory and neuroprotective effect on these other cells. This is still controversial.
In the FREEDOMS trial (2010),[80] 1 272 subjects were randomised to receive either 0.5 or 1.25 mg fingolimod daily, or placebo, for a period of 2 years. The ARR in each of the 3 groups was 0.18, 0.16 and 0.40, respectively and the probability of 3-month confirmed EDSS progression was 17.7%, 16.6% and 24.1%, respectively. The active groups also had less new T2 lesions, contrast-enhancing lesions and reduction in brain volume.
In the TRANSFORMS trial (2012),[81] 1 292 subjects were randomised to receive either 0.5 or 1.25 mg/day fingolimod, or IFN-beta-1a (30 µg Avonex) IMI weekly for 1 year. The ARR was 0.16, 0.20 and 0.33, respectively. There was no difference in the confirmed disability progression.
6.3.3.1 Risks of fingolimod1821
In the TRANSFORM trial, 1 patient developed a fatal herpes zoster infection after exposure to chicken pox and 1 patient died of herpes simplex encephalitis. Both of these patients were on the 1.25 mg dose and both recently received steroids. A mild increase in lower respiratory infections and herpes virus infections has also been reported.
Patients may develop a bradycardia after the first dose of fingolimod. This occurs in 1 - 3% of patients and is usually asymptomatic. Second-degree atrioventricular (AV) block occurs in <1% of patients. A mild increase in systolic and diastolic blood pressure may occur.
One death was reported in December 2011. This occurred after the 6-hour observation period, but within 24 hours after the first dose of fingolimod. The FDA concluded that there was no clear evidence that fingolimod played a role in the death. However, a link could not be ruled out. Further analysis of Holter electrocardiography (ECG) data revealed that a second decrease in heart rate occurs 12 - 20 hours post-dose, in part related to the circadian rhythm. The FDA has consequently made the following recommendations (http://www.fda.gov/Drugs/DrugSafety/ucm303192.htm) (Please refer to the package insert for full details):
• Hourly pulse and blood pressure monitoring for the first 6 hours
• ECG before the first dose and at the end of the 6 hours
• Patients who develop a heart rate of <45 bpm at the end of the 6 hours or a new second degree (or higher) heart block, must be observed until resolution of this finding
• Patients with symptomatic bradycardia must be observed with continuous ECG until resolution. If pharmacological intervention is required they should be observed overnight and the first dose procedure should be repeated with the second dose
• Certain patients at high risk (i.e higher risk of bradycardia or heart block due co-existing medical condition or medication, those with a prolonged QTc interval at baseline or during the observation period, or receiving drugs with known risk of torsades de pointes) should be observed with continuous ECG overnight.
If fingolimod is discontinued for >2 weeks, the first dose procedure should be performed on reintroduction.
Macular oedema occurred in <1% of patients and was mostly asymptomatic; it resolved after discontinuation of fingolimod.
Skin cancer (basal-cell carcinoma and melanoma) and breast cancer were reported more frequently in the fingolimod arms, compared with Avonex, in the TRANSFORMS trial. However, there was no difference in frequency between the active arms and placebo arm in the FREEDOMS trial. Further long-term follow-up has been recommended
Patients receiving fingolimod developed a mild dose-dependent decrease in lung function over the first month. There was also a decrease from baseline in the circulating lymphocyte count - 73% (0.5 mg dose) and 76% (1.25 mg dose) in the FREEDOMS trial. This occurred in the first month and then remained stable. Patients receiving fingolimod frequently develop elevated liver enzymes, but this is asymptomatic and resolves even when fingolimod is continued.
6.3.4 Recommended monitoring
6.3.4.1 Baseline evaluations to be performed before therapy initiation
• Liver functions, total and differential white cell count and varicella-zoster serology. Immunisation 1 month before initiating fingolimod should be considered if the serology is negative.
• In the case of significant ECG abnormalities, referral to a cardiologist is recommended.
• Baseline ophthalmological evaluation is indicated in patients with visual symptoms, or patients at higher risk of macular oedema.
6.3.4.2 Medication washout
• Patients should not have received immunosuppressive treatment or natalizumab for 3 months before initiation of fingolimod.
• For cyclophosphamide and mitoxantrone, the washout period should be 6 months.
6.3.4.3 With the first dose
• Vital signs must be monitored for the first 6 hours.
• An ECG should be done before the first dose and at 6 hours. Specific cases may require overnight observation (see above).
6.3.4.4 Monitoring on treatment
• White cell counts, aminotransferase and bilirubin levels should be repeated at 3 months and then at least annually.
• Ophthalmological evaluation should be repeated at 3 - 6 months, and thereafter when new visual symptoms occur.
• Annual dermatological evaluation is recommended.[82]
6.3.5 Recommendations
The MS Advisory Board recommends the following regarding the choice of second-tier DMT (Fig. 2):
6.3.5.1 Fingolimod
Fingolimod is expected to be approved as first-tier therapy by the MCC. However, we recommend that it be reserved as second-tier therapy, except in those patients with highly-active disease at diagnosis.
In patients with significant bradyarrhythmias or macular pathology, natalizumab may be a preferable choice.
6.3.5.2 Natalizumab
Natalizumab should be avoided in patients with positive JCV serology. The risk of PML becomes significant after 2 years, especially in patients who had prior exposure to immunosuppressive medication, estimated at 0.8%. However, discontinuation at 2 years becomes problematic since it may result in rebound disease. In these patients fingolimod would thus be preferable.
6.3.5.3 Mitoxantrone
• The maximum lifetime dose of mitoxantrone is 96 - 140 mg/ m2, thus 2 years of uninterrupted treatment. Mitoxantrone is consequently not a long-term option.
• Mitoxantrone has significant potential adverse effects, specifically cardiomyopathy - which requires careful monitoring - and TRAL. The risk of leukaemia is significant (0.8%), and may occur up to 5 years after exposure.
• Previous use of mitoxantrone increases the risk of developing PML by 3 - 4 fold in patients who are later treated with natalizumab.
• Mitoxantrone should thus be reserved as a 'third-tier' therapy (Fig. 2).
• Since fingolimod and natalizumab will now be available, we recommend that mitoxantrone only be considered in the following situations:
1. Continued disease activity despite treatment trials with both fingolimod and natalizumab.
2. Continued disease activity while receiving fingolimod, where the JCV serology is positive.
3. Induction therapy (monthly infusions for 6 months) in very highly-active disease.
4. Patients with SPMS with an EDSS of <7 and who failed on other DMTs.
7. Monitoring of already diagnosed patients
Monitoring of patients with an illness such as MS is essential. It is recommended that patients are at least twice per year.[83,84] Patients need to be assessed for progression of disease, problems related to the disease and the effect of therapy on the disease.
Strategies for monitoring patients and assessing therapies for MS include clinical measures, neuro-imaging measures, disability scales and quality-of-life (QoL) measures?5-
Progression may be defined as a function of neuropsychological tests,[86] and it is advisable to perform them at the time of diagnosis to have a baseline value for comparison on subsequent evaluations. The most frequent scale to evaluate MS progression is the EDSS.[87] It is also recommended that neurologists administer the MSFC test if the clinicians are trained in its administration, as this is a more accurate measurement of cognitive impairment.[84]
Neuro-imaging involves MRI and all its modalities. It has set the standard for evaluating the effectiveness of treatment, disease activity, disease burden and disease type. It may differentiate the transition to SPMS from treatment failure. MRI can detect a large amount of subclinical disease activity, which is a major advantage of over clinical monitoring; however, MRI findings alone may not be helpful in predicting disability in an individual patient.[88]
The administration of an MRI is considered mandatory in cases of suspected lack of efficacy.[84,89] MRI needs to be done if there is a change in the patient's condition. The MRI must be done with recognised protocols and include Gd to minimise missing relapse activity. In MS, the number of detectable active lesions increases with Gd enhancement, which most likely correlates with pathological activity.
A combination of both clinical and MRI measures gives better clarification as to the patient's progression and response to treatment. MRI scans appear to be most useful in the first year after the start of MS, to help gauge disease activity and to aid diagnosis. Later in the disease, repeat MRI scans are performed for investigation of new symptoms or assess the level of inflammation if treatment change is contemplated.[90]
7.1 Monitoring suboptimal response to therapy
Treatment failure has been defined as a function of the frequency of relapses, but it must be evaluated in relation to the frequency of relapses before treatment.
A combination of both clinical and MRI measures needs to be used for assessment of an adequate response to therapy.
The immunological steady state associated with a full therapeutic effect is believed to occur from 6 months to 1 year after patients begin IFN-beta or GA therapy. An MRI scan to evaluate active lesions should be performed during the first 6 - 12 months of therapy. In patients with >2 active lesions and with clinical activity (i.e relapses or an increase of disability) after 6 months from initiating therapy, are likely experiencing suboptimal or partial therapeutic responses. [91] The occurrence of >1 relapse per year, or the failure of a given treatment to reduce the relapse rates from pre-treatment levels is considered clinical evidence of a suboptimal therapeutic response.[92]
If the response is not sufficient, a change in therapy is indicated.
Close clinical monitoring is required in the absence of clinical activity despite new MRI activity. In the case of the appearance of relapses or an increase of disability, a change of therapy also needs to be considered.[84]
A change in therapy can be within tier, escalation to a higher tier, or to combination therapy. The current recommendations are to start with immunomodulatory agents as first-line therapy (IFNs and GA) and then advance treatment if there is an inadequate response until the disease is effectively controlled.[92] The first step may be to switch between therapeutic first-tier drugs and then to potentially more effective second-tier drugs such as the monoclonal antibody (mAb) natalizumab, fingolimod (not yet approved in SA), or mitoxantrone. The decision regarding therapeutic escalation should be made as soon as possible after detection of treatment failure to prevent irreversible neurological impairment. If previous treatments are not effective and MRI scans continue to reveal inflammatory activity, or if the patient continues to relapse, a third level of treatment needs to be considered, including drugs not approved for the treatment of MS (off-label indications), such as high doses of rituximab[93] or alemtuzumab,[94] cyclophosphamide[95] or bone marrow transplant.
7.2 Considerations for discontinuing therapies
Refer to section 5.2.
8. Potential new targets and therapies
These include anti-T- and anti-B-cell therapies, T-regulatory enhancement, immune regulation (that enhance T-regulatory function), reduced permeability of the BBB, prevention of transgression of the BBB, targeting key members of the inflammatory cascade (cytokine therapies), immune tolerance with putative auto-antigens, neuroprotection to prevent neuronal loss, growth factors to promote remyelination and augment regenerative repair.
We have been recently blessed with a plethora of trials and potential medications to attack these various targets; it is therefore necessary to re-address the current treatment algorithm to allow exit strategies from medications that are not working and the introduction of new second-line therapies that are mostly new biologicals that do not come without their inherent risks and costs.
8.1 Potential new therapies (phase III trials)
8.1.1. Biological therapies
8.1.1.1 mAbs
8.1.1.1.1 Anti-CD52 (alemtuzumab, Campath-1H)
Alemtuzumab is a humanised IgG1 mAb that binds to CD52 on leucocytes, including T- and B-cells, natural killer cells, monocytes and macrophages, depleting these cells. It also stabilises the BBB and 'resets' the immune system. On immune reconstitution there is a reduced proliferation of autoreactive T-cells, as well as an overshoot of B-cells by 50%, indicating a fundamental shift in immune status.
Anti-CD52 is an annual infusion over 3 - 5 days. It was initially tried in SPMS patients with improvement on serial scans, but no clinical benefit, and was then tried in early RRMS patients in comparison with Rebif, a principle IFN first-line therapy.[96]
There was an improvement in the EDSS disability score compared with a deterioration in the Rebif group. An 80% reduction in relapses v. Rebif over the same period and a dramatic reduction in MRI activity were observed. Patients (30%) developed an autoimmune thyroiditis and idiopathic thrombocytopenic purpura (ITP) developed in 6 subjects with death in 1 due to intracranial haemorrhage as a result of poor clinical vigilance.[94]
With delayed B-cell reconstitution and then overproduction, many autoimmune diseases can be expected i.e. ITP, haemolytic anaemia, immune-mediated neutropenia, etc. and this seems to be related to the genetically predetermined baseline IL-21 levels. By measuring a person's IL-21 levels, their likelihood of developing autoimmunity after alemtuzumab therapy can be predicted.
There is also a higher rate of infection in particular recrudescence of herpes infections. Two large phase III trials are underway in treatment-naive and failed first-line therapy patients.
8.1.1.1.2 Anti-CD20 (rituximab, ocrelizumab and ofatumumab)
These agents target CD20 expressed on mature B-cells, but not on plasma cells that results in a transient (6 month) depletion of >60% of circulating B-cells. The repopulation of B-cells occurs via the marrow with naive B-cells.[93]
Phase II trials showed a rapid sustained reduction of MRI activity over a 72-week period and phase III trials are underway for RRMS.[97,98]
In PPMS there was a delay in time to confirmed disease progression and a reduction in T2 lesions on MRI in subjects aged <51 years with Gd-enhancing lesions on MRI.
PML has been reported in patients treated for lymphoma and rheumatological disorders. Ocrelizumab use has been suspended in SLE and rheumatoid trial patients after serious and fatal opportunistic infections.
8.1.1.1.3 Anti-CD25 (daclizumab)
Daclizumab has been used for the prevention of transplant rejection and blocks CD25, which forms part of the high-affinity IL-2 receptor. CD25 is up-regulated in activated T-cells. Blocking CD25 reduces T-cell proliferation. Its therapeutic effect is not well known. It inhibits the survival of CD4, CD25, forkhead box (FOX) P3 and T-regulatory cells.[99]
Phase II trials performed in patients who failed IFN-beta therapy showed stabilisation of disease and decreased Gd-enhancing lesions on MRI. Trials comparing IFN-beta alone with patients receiving IFN-beta and daclizumab showed a greater decrease in MS disease progression parameters in the combination therapy cohort.[100]
Phase III trials to evaluate therapeutic and safety are currently underway.[101]
8.1.1.2 Oral agents
All current agents available in SA require SC injections and IMIs on a frequent basis with associated site reactions, infections, lipoatrophy and systemic side-effects. The previously mentioned biological agents require repeated parenteral therapy with major potential side-effects.
This lays open the need for safe and efficacious oral therapies with fewer side-effects. At the time of writing this guideline, 2 agents have been released with 1 agent (cladribine) already withdrawn due to benefit-risk ratios not being met. The second agent, fingolimod, has not been released in SA but submission has been made to the MCC, thus it will be mentioned under new therapies.
There are 4 promising oral agents in phase III trials viz. BG-12 (dimethyl fumarate), fingolimod, laquinamod and teriflunomide.
8.1.1.2.1 BG-12
Also known as Fumaderm, this is an agent commonly used in the treatment of psoriasis. An advantage over other agents is that there is extensive clinical experience with the drug and its safety profile.
Fumarate functions via the NF-E2 transcription pathway that controls phase II detoxifying enzyme gene expression, which plays a role in oxidative stress-induced neuronal damage. Activation of this pathway protects the integrity of the BBB as well as myelin integrity by inhibiting pro-inflammatory cytokines and adhesion molecules.
In phase II trials BG-12 reduced Gd-enhancing lesions by 69% and the ARR. Phase III trials were drawing to an end as this guideline was being written, with promising results; and they were performed as head-to-head trials with GA.[102]
The low cost of the drug and vast clinical experience make this drug a promising future agent.
8.1.1.2.2 Fingolimod (Gilenya)
This agent was recently released in the US and Russia as a first-line therapy with second-line therapy registration in Europe and the UK.
It is a structural analogue of S1P, which plays an important role in lymphocyte migration from lymph nodes to the periphery by internalising the sphingosine receptor, trapping the lymphocyte in the peripheral node. It readily crosses the BBB and interacts with central S1P receptors. It can promote remyelination in animal models. It down-regulates inflammatory genes, adhesion molecules and metalloproteinases, thereby maintaining the integrity of the BBB.
Phase III trials showed a 52% reduction in relapse rate compared with IFN (38%). Reduction in MRI lesion load and activity was also demonstrated. Mild side-effects were reported in >90% of patients, with the more serious being bradycardia and AV block as a first dose effect with deaths being reported in predisposed cardiac patients. Other notable side-effects included macular oedema, increased herpes recrudescence and hypertension. An increased risk of malignancies is being monitored with prolonged use.
The drug, as mentioned above, is available overseas and is awaiting registration in SA.
8.1.1.2.3 Laquinamod
Laquinamod is derived from linomide, which was initially trialled in MS but withdrawn due to side-effects. It is an immunomodulatory agent that induces the release of TGF, a shift in immune response to a TH2 profile, reduced leucocyte infiltration of CNS and shift of MBP-specific cells to a TH2 pattern.
Phase II trials showed a reduction in MRI lesions by 44% compared with placebo. In the open-label extension phase, the proportion of patients free from Gd-enhancing lesions increased from 31% at baseline to 47%. Adverse events included a mild liver transaminitis.[103]
Phase III trials are underway against Avonex v. placebo.[104]
8.1.1.2.4 Teriflunomide
Teriflunomide is the active metabolite of leflunomide, which is used in the treatment of rheumatoid arthritis. It inhibits the mitochondrial enzyme dihydroorotate dehydrogenase, which plays a role in pyrimidine synthesis interfering with T- and B-cell proliferation. Teriflunomide also inhibits tumour necrosis factor (TNF)-alpha and IL-2.
Phase II trials comparing 2 doses showed a marked decrease in the number of active and new lesions on MRI. EDSS deterioration and relapses were decreased. Extension studies performed over 8 years showed good tolerance and side-effect profiles.[105]
Recently released data from a phase III trial showed a 32% reduction in ARR. Risk of disability progression was reduced by 29%. The drug was well tolerated with withdrawal due to side-effects averaging 10% and no reported deaths. There are currently several phase III trials assessing combination therapy with IFN-beta and GA.[106]
8.1.1.2.5 Cladribine
This drug was withdrawn after release in Australia and Russia and will therefore not be discussed because it is no longer available.
8.1.1.3 Conclusion
Not all patients respond to IFN-beta or GA. The escalation therapies of mitoxantrone and natalizumab have come under scrutiny, with up to a 1% chance of haematological malignancies in the former, and with the risk of PML after 2 years, if the patient is JCV-positive, in the latter.
The newer medications appear more efficacious but are more toxic and long-term safety data are not available. However, many patients are willing to take the risk early on to prevent further disability down the line, therefore the option of switching from ineffective first-line therapies to the newer agents has to be offered to the patient, necessitating the development of this revised treatment guideline.
9. Symptomatic treatment
Patients diagnosed with MS suffer great morbidity due to the symptoms associated with this disease. Patients on DMT, as well as those who do not qualify for disease-modifying medication, can have significant improvement in QoL when at least some of these symptoms are eliminated or at least reduced.
The first principle in symptomatic management is to avoid the use of drugs that will worsen symptoms. Examples include: (i) tricyclic antidepressants that improve depression and nocturnal bladder control, but worsen fatigue; (ii) selective serotonin reuptake inhibitors (SSRIs), which improve depression, but worsen sexual dysfunction; and (iii) baclofen that can reduce spasticity, but can induce weakness.
9.1 Fatigue
Fatigue is a well-known symptom of MS. However, it is sometimes difficult to distinguish daytime fatigue from depression. Careful examination of the course of fatigue during the day can help to distinguish between fatigue and depression. Worsening of fatigue as the day progresses and improvement of fatigue with rest is an indication that the patient suffers from fatigue rather than from depression.
The fatigue can be primary to MS or secondary to MS symptoms. The first step in managing fatigue is to look for secondary causes of daytime fatigue. Sleep disturbances, reduced mobility, spasticity, anxiety, pain, depression, infections, drugs and other medical conditions should be managed before embarking on drug management of fatigue.
An occupational therapy assessment with the aim of optimising energy conservation and advice on planning the patient's day can be very useful in the management of fatigue.
Modafinil can be very useful in managing fatigue. This drug works by selectively increasing neuronal activity in the hypothalamus and by activating tuberomammillary nucleus neurons, which release histamine. It also activates hypothalamic neurons, which release orexins/hypocretins. The recommended dosage is 100 - 400 mg/day, starting with 100 mg in the morning, increasing as necessary to a morning and midday dose. Nervousness, insomnia, headache, anorexia and a dry mouth are among the more common side-effects.
Amantadine is useful in some patients.[107] This drug, more commonly used in Parkinson's disease, seems to work either by inducing dopamine release or by decreasing dopamine uptake. It is also a weak N-methyl-D-aspartic acid (NMDA) receptor antagonist. The more common side-effects include nausea, dizziness, insomnia and blurry vision. This drug, if co-prescribed with memantine, can produce hallucinogenic side-effects. The suggested dose is 100 -200 mg/day, with an initiation dose of 100 mg in the morning and a maximum dose of 400 mg/day.
The newer-generation antidepressants can improve daytime fatigue, even in the absence of underlying depression. Some anecdotal evidence exists that both SSRIs and serotonin and noradrenalin-reuptake inhibitors (SNRIs) can improve fatigue in patients with MS. These drugs are particularly useful where fatigue, underlying depression and anxiety co-exist. Among the most commonly used drugs in this class is a morning dose of 20 mg fluoxetine, 50 mg sertraline or 75 mg venlafaxine. Side-effects include insomnia, nausea and sexual dysfunction.
There is also some evidence that methylphenidate may be beneficial. L-carnitine at a dosage of 1 g twice daily has been suggested to help with fatigue.
Methylphenidate can be used in doses of up to 54 mg/day; drug holidays are suggested.
9.2 Cognition
In the past it was believed that MS rarely affects cognition. This is no longer considered to be the case and it has become increasingly apparent that cognition is not spared by this condition.
Glucocorticoid therapy often gives temporary improvement of cognition. Unfortunately, this is a relatively short-lived effect.
Cil,[108] as with memantine,[109] have yielded mixed results. However, some studies showed slight improvements in memory. Care should be taken when co-prescribing memantine with amantadine due to the hallucinogenic side-effects.
In patients with cognitive symptoms co-existing with depression or fatigue, one should primarily treat the depression or fatigue.
9.3 Affective disorders
Depression is commonly seen in patients with MS. This can present as a typical depressive episode, but poor cognition, lethargy and sexual dysfunction as presenting symptoms of depression, are not uncommon.
In patients where depression presents as a primary problem, with fatigue as an associated feature, SSRIs[110] and SNRIs are useful first-line agents. When prescribing these drugs, one should be vigilant about symptoms of nausea, insomnia and sexual dysfunction as side-effects of these drugs. Among the more commonly used agents are fluoxetine (20 mg), citalopram (20 mg) and sertraline (50 mg).
Where central neuropathic pain and depression co-exist, one should consider prescribing duloxetine at a dose of 60 mg/day.[110]
This SNRI is registered for peripheral neuropathic pain, but is very useful in the treatment of central neuropathic pain as well as fatigue. The side-effects are similar to that of the SSRIs and the SNRIs.
In patients with depression and underlying spastic bladder or insomnia, the choice of a lower dosage tricyclic antidepressant (TCA) can be extremely useful. The common side-effects are dry mouth, constipation and daytime fatigue, among others. Amitriptyline at 10 - 25 mg at night is most commonly used.
Abulia, although an uncommon symptom, can respond well to dopamine-enhancing drugs. There is some evidence that amantadine, bromocriptine, pramipexole and ropinderole can improve these symptoms.
9.4 Spasticity
The treatment of spasticity in MS can significantly improve the quality of life of the patient. The balance between reducing spasticity and inducing weakness needs to be considered carefully.
The first step is to reduce or, if possible, eliminate underlying conditions that intensify spasticity. Among others, these include pain, constipation, bladder infections fear and anxiety.
Generally, the first-line approach would be regular physiotherapy. Hydrotherapy and dry needling are avenues that the physiotherapist can use in addition to the standard measures to reduce spasticity.
When considering drug therapy for spasticity, one must differentiate between generalised spasticity and disabling focal spasticity.
With disabling focal spasticity, botulinum toxin can be used focally, thus avoiding the systemic side-effects of other drugs.[111] In MS this is particularly useful in adductor spasm, both in the severely disabled patient to improve perineal hygiene, and in the less disabled patient, to improve gait.
The 2 first-line agents for generalised spasticity are baclofen and benzodiazepines. Benzodiazepines are particularly useful in painful or painless nocturnal muscular spasms that disturb sleep. Diazepam (5 - 10 mg at night) is a simple solution. Unfortunately, tolerance can become an issue with benzodiazepines. These drugs are difficult to use in the day as they are sedating and intensify fatigue. However, in a patient with insomnia, anxiety and nocturnal spasms, they can be extremely useful.
Baclofen is probably more useful if a reduction in spasticity during the day as well as at night is required.[112] It is a gamma-aminobutyric acid (GABA)-B receptor agonist and works at the spinal and supra-spinal levels. It inhibits spinal interneurons through postsynaptic hyperpolarisation. This drug is contraindicated in epilepsy and severe renal impairment. The most common side-effects are exhausting muscle weakness, ataxia, tremor, dizziness confusion and orthostatic hypotension.
The general approach is to start low and increase slowly until a satisfactory result has been obtained, starting at 5 - 10 mg 3 times per day. The optimal dosage is usually 30 - 75 mg/day. In patients who either cannot tolerate the maximum oral dosage or need more to relieve the spasticity, intrathecal baclofen is an extremely useful alternative. A test dosage is required; thereafter, an intrathecal baclofen pump can be inserted. In MS this is usually a lumbar intrathecal pump, but with selected patients a higher placement is possible.
Dantrolene can also be successfully used for spasticity. Gabapentin[113] and pregabalin can be tried for reducing spasticity. These agents are particularly useful in patients who have concomitant neuropathic pain.
9.5 Bladder dysfunction
Bladder dysfunction is an emotionally disturbing and socially-isolating problem in patients with MS. The most common symptoms are storage problems (i.e. urgency, urge incontinence and frequency). Voiding issues can also occur (i.e. slow stream, hesitancy, straining and incomplete voiding). A combination of both sets of symptoms commonly occurs.
Although the predominant underlying symptom can be treated, there is no doubt that a urological assessment with an urodynamic assessment will optimise the management of the patient's symptoms.
Often the first step in managing a patient who has a combination of insomnia with nocturnal frequency can simply be addressed by an anticholinergic agent. Where the frequency is mainly a nocturnal problem, a TCA with anticholinergic activity is a simple first measure (e.g 10 - 25 mg amitriptyline/imipramine at night).
In cases where failure-to-store is the primary disorder, oxybutynin, tolterodine or trospium are useful. Alternatively, darifenacin or solifenacin can be used.[114] Botulinum toxin can be injected for detrusor hyperreflexia.[115] With failure to empty, intermittent catheterisation is probably best, but occasionally tamsulosin can be helpful.
Patients with recurrent urinary tract infections can benefit from long-term urinary antiseptics.
There is some evidence that cannabinoids improve overactive bladder symptoms in patients with MS.
9.6 Sexual dysfunction
MS is a disease that affects young patients and sexual dysfunction is a common and distressing problem. In patients with diminished sexual desire the best approach is probably counselling and psychotherapy. The patient's drug regimen also needs to be reviewed as SSRIs can cause diminished sexual desire.
In male patients with poor or absent erections, phosphodiesterase inhibitors such as sildenafil (25 - 100 mg)[116] or tadalafil (5 -10 mg) can induce erections in most patients. In patients who fail on these options, intracavernous injections of a prostadil can be considered.
In women where sexual desire is normal, but enjoyment is not, there are less options. A first step is to use vaginal lubrication.
9.7 Central neuropathic pain
Neuropathic pain is a disabling feature of MS. It exacerbates spasticity and worsens insomnia and depression. In patients with MS related to trigeminal neuralgia, carbamazepine is the first-line drug of choice.[117] The dosage varies from 100 - 400 mg 3 times per day to 200 - 600 mg twice per day depending on whether the immediate or controlled release preparation is used.
Pregabalin works presynaptically on overactive calcium channels and can reduce the symptoms of neuropathic pain. The therapeutic dosage is usually 75 - 150 mg twice daily, starting slowly as drowsiness and dizziness are common side-effects.
Duloxetine works centrally on the ascending and descending pain pathways by modulating both noradrenalin and serotonin. Dosages of 60 - 90 mg in the morning can help the symptoms of central neuropathic pain. In patients with co-existent fatigue and depression, this is particularly useful. Nausea and insomnia are among the more common side-effects. There is some anecdotal evidence that cannabinoids help to reduce MS central neuropathic pain.
9.8 Other phenomena
• Foot drop. Splinting, whether fixed or dynamic, can dramatically improve a patient's gait. The use of a drop-foot stimulator and applying a stimulator at the knee can also be useful.
• Ataxia. Physiotherapy with the appropriate walking aids can help the patient.
• Tremor. Patients with a rubral tremor are extremely difficult to treat. Standard tremor medications such as trihexylphenidyl, biperidone or orphenadrine can be tried, but are usually unhelpful. Occasionally patients will improve with isoniazid.[118]
• Visual loss. Little can be done to help patients with profound visual loss, but with milder loss, simply improving lighting and using larger fonts on computers can help.
• Impaired gait causes major disability in MS patients. Dalfampridine (4-aminopyridine) is a potassium channel blocker. The extended-release product has shown a 25% increase in walking speed in 37% of patients.[119] Side-effects include seizures, acute encephalopathy and confusion.
10. The role of physiotherapy
The role of the physiotherapist in the management of MS patients is a varied and important one and should not be restricted to patients who have already lost important functions. Early referral is desirable to commence appropriate management and allow a patient to reach their full potential while limiting secondary complications. It is recommended that a physiotherapist has continuous involvement with an MS patient, with varying intensity throughout the course of the disease.[120,121,122]
Long-term physiotherapy on a regular basis provides 2 major avenues to counteract the motor dysfunction caused by MS. It minimises disability resulting from postural deformity and disuse atrophy, and maximises functional abilities at each stage of the disease.[123] A course of physiotherapy has been shown to improve mobility, subjective well-being, and mood in chronic MS patients compared with no treatment.[124]
Referral to physiotherapy should be considered for:[122,124]
• improving balance and walking difficulties
• reducing muscle spasms and stiffness
• increasing strength
• retraining normal patterns of movement
• increasing energy levels
• reducing bladder or bowel dysfunction
• improving mood and a sense of well-being
• reducing the risk of falls
• advice regarding walking aids and equipment to make the home/ working environment safer
• education regarding MS and its symptoms.
The fundamentals of rehabilitation in the early stages of the disease revolve around encouraging activity while minimising the impact of fatigue as well as daily stretching in combination with activities that stimulate balance and posture. Active strengthening and cardiovascular fitness are promoted within boundaries to prevent over-fatigue of the muscles and the patient as a whole.[120,121]
As motor deficits become more marked and activity decreases, a thorough assessment of movement is performed to identify deficits in execution and control of voluntary movement, as well as function and activities of daily living. Goal-setting becomes based on the level of disability and concurrent stability. Symptoms that require specific management will include spasticity, ataxia/loss of control of voluntary movement, fatigue, postural abnormalities, and loss or deterioration of bladder and bowel control. Full-time use of a wheelchair must be avoided for as long as possible, as the inevitable reduction in activity and increased use of sitting posture usually leads to alteration in patterns of spasticity and movement. It is essential to maintain standing, weight-bearing and walking for as long as possible, but constantly weigh up the benefits v. the need to protect against fatigue.
The immobile patient requires special management, including respiratory training and re-education, circulatory exercises done actively or passively, contracture prevention through movement of all joints through full range of motion, as well as correct positioning. Adequate pressure care must be maintained through correct positioning and regular turning.[120,123]
Throughout the rehabilitation process, the physiotherapist will not only play an important part in the treatment and management of symptoms, but will also fulfil an educative and supportive role for the patient, family and caregivers by passing on information, advice and handling techniques[120,122-124]
10.1 Occupational therapy
10.1.1 Definition
'Occupational therapists (OTs) are concerned with occupational performance, i.e. the ability to carry out activity that is both relevant and meaningful, while participating in society through the fulfilment of chosen roles and routines.[125] Occupation, in this context, is defined as everything people do day-to-day to occupy themselves, including enjoying life (leisure), looking after themselves (self-care), and contributing to society (productivity). OTs focus on the dynamic interaction between the individual, their occupations and their environment to promote independence and QoL.[126]
OTs are believed to play a unique role among people with MS who experience limitations in their ability to function due to the range and complexity of physical and cognitive changes in their health condition. The key areas addressed by an OT would include activity of daily living (ADL) retraining, vocational rehabilitation, leisure activity promotion, environmental adaptations, family/caregiver training, assistive device provision and fatigue management.
10.1.2 ADLs
A significant proportion of people with MS may eventually experience limitations in their ability to undertake a variety of activities needed to live independently, such as looking after themselves, doing shopping and performing housework. Collectively these activities are referred to as ADLs. Occupational therapists are usually the professional group with the most expertise in, and who lead in, the rehabilitation of ADL.[127]
10.1.3 Vocational activities: Employment and education
It is important to attempt to minimise the impact of MS on an individual's productivity, because studying and employment have many health benefits and promote self-esteem, social interaction, financial independence and a valued position in society. The importance of vocational rehabilitation services for people with MS at all stages, particularly in the early stages, was emphasised many years ago[128] and has been reiterated recently.[127]
10.1.4 Leisure and social interaction
Leisure activities are those which give balance to the stresses and strains of everyday life, and provide opportunities for social interaction. Impairments and disabilities that may affect work will usually also affect leisure. The importance of leisure as an area worthy of attention by specialist rehabilitation services is being increasingly recognised, but it is often only addressed in the community and seems a lower priority compared with self-care and productivity.[127]
10.1.5 Equipment, adaptations and family/carer training
The consequences of any disease are influenced greatly by a person's home, work and social environments. The effectiveness of providing assistive equipment has been demonstrated in a randomised control trial in the US,[129] OTs are specialists in assessing and adapting an individual's environment to meet their changing needs.
The physical environment also includes family members and/ or caregivers, who may provide hands-on assistance, ensure safety, provide prompts, or merely provide the reassurance needed to allow an individual to perform an activity independently. OTs play a crucial role in training and educating family members and carers to assist people with MS appropriately.[127]
10.1.6 Fatigue management/energy conservation
Fatigue management strategies can be very effective when fatigue is identified as a major limiting factor in the performance of daily activities and the fulfilment of chosen roles and interests. 'Fatigue management is a process that increases understanding of the primary and secondary factors, which contribute to and exacerbate fatigue. Then, through education and adaptation, individuals learn to optimise their ability to function within the context of fatigue through goal setting and the use of energy conservation strategies'.[13°-'Such strategies include work simplification, ergonomics and labour-saving equipment; prioritisation, time management and planning; a balance between rest/relaxation and exercise; graded activity with increasing exercise tolerance; role adjustment and assistance from community services.[134]
The role of an OT is well-recognised in the management of chronic, persistent fatigue in MS.[133-136]
10.1.7 Summary
In summary, OTs have a valuable role to play among people living with MS. Their primary role is to maximise and maintain independence and participation, ADLs, leisure tasks, work, and life roles specific to each individual, as the disease progresses. Interventions may include education, carer/family training, provision of equipment and aids, adaptation of environments, ADL retraining and fatigue management.
References
1. Compston A, Coles A. Multiple sclerosis. Lancet 2002;359(9313):1221-1231. [http://dx.doi. org/10.1016/S0140-6736(02)08220-X] [ Links ]
2. Kurtzke JF. Epidemiologic evidence for multiple sclerosis as an infection. Clin Micro Rev 1993;6(4):382-427. [ Links ]
3. Okuda DT, Mowry EM, Beheshtian A, et al. Incidental MRI anomalies suggestive of multiple sclerosis: The radiologically isolated syndrome. Neurology 2009;72(9):800-805. [http://dx.doi.org/10.1212/01.wnl.0000335764.14513.1a] [ Links ]
4. Barkhof F, Filippi M, Miller DH, et al. Comparison of MRI criteria at first presentation to predict conversion to clinically definite multiple sclerosis. Brain 1997;120(11):2059-2069. [http://dx.doi.org/10.1093/brain/120.11.2059] [ Links ]
5. Tintoré M, Rovisa A, Rio J, et al. New diagnostic criteria for Multiple sclerosis: Application in first demyelinating episode. Neurology 2003;60(1):27-30. [http://dx.doi.org/10.1212/WNL.60.L27] [ Links ]
6. Lebrun C, Bensa C, Debouverie, M et al. Association between clinical conversion to multiple sclerosis in radiologically isolated syndrome and magnetic resonance imaging, cerebrospinal fluid and visual evoked potential: Follow-up of 70 patients. Arch Neurol 2009;66(7):841-846. [http://dx.doi.org/10.1001/archneurol.2009.119] [ Links ]
7. Siva S, Saip S, Altintas A, et al. Multiple sclerosis risk in radiologically uncovered asymptomatic possible inflammatory-demyelinating disease. Mult Scler 2009;15(8):918-927. [http://dx.doi.org/10.1177/1352458509106214] [ Links ]
8. O'Riordan JI, Thompson AJ, Kingsley DPE, et al. The prognostic value of brain MRI on clinically isolated syndromes of the CNS. A 10-year follow-up. Brain 1998;121(3):495-503. [http://dx.doi.org/10.1093/brain/121.3.495] [ Links ]
9. Masjuan J, Alvarez-Cermeno JC, Garcia-Barragan N, et al. Clinically isolated syndromes: A new oligoclonal band test accurately predicts conversion to MS. Neurology 2006;66(4):576-578. [http:// dx.doi.org/10.1212/01.wnl.0000198253.35119.83] [ Links ]
10. Pelayo R, Montalban X, Minoves T, et al. Do multimodal evoked potentials add information to MRI in clinically isolated syndrome? Mult Scler 2010;16(1):55-61. [http://dx.doi.org/10.1177/1352458509352666] [ Links ]
11. Lublin FD, Reingold SC and National Multiple Sclerosis Society (USA) Advisory Committee on Clinical Trials of New Agents in Multiple Sclerosis. Defining the clinical course of multiple sclerosis: Results of an international survey. Neurology 1996;46(4):907-911. [http://dx.doi.org/10.1212%2FWNL.46.4.907] [ Links ]
12. Thompson AJ, Polman CH, Miller DH, et al. Primary progressive multiple sclerosis. Brain 1997;120(6):1085-1096. [http://dx.doi.org/10.1093/brain/120.6.1085] [ Links ]
13. Cree BA, Khan O, Bourdette D, et al. Clinical characteristics of African Americans vs Caucasian Americans with multiple sclerosis. Neurology 2004;63(11):2039-2045. [http://dx.doi.org/10.1212/01.WNL.0000145762.60562.5D] [ Links ]
14. Mowry EM, Pesic M, Grimes B, et al. Demyelinating events in early multiple sclerosis have inherent severity and recovery. Neurology 2009;72(7):602-608. [http://dx.doi.org/10.1212/01.wnl.0000342458.39625.91] [ Links ]
15. Link H, Huang YM. Oligoclonal bands in multiple sclerosis cerebrospinal fluid: An update on methodology and clinical usefulness. J Neuroimmunol 2006;180(1):17-28. [http://dx.doi.org/10.1016/j.jneuroim.2006.07.006] [ Links ]
16. Schumaker G, Beebe G, Kibler R, et al. Problems of experimental trials of therapy in multiple sclerosis: Report by the panel on the evaluation of experimental trials of therapy in multiple sclerosis. Ann NY Acad Sci 1965;122:552-568. [http://dx.doi.org/10.1111/j.1749-6632.1965.tb20235.x] [ Links ]
17. Poser C, Paty D, Schienberg L, et al. New diagnostic criteria for multiple sclerosis: Guidelines for research protocols. Ann Neurol 1983;13(3):227-231. [http://dx.doi.org/10.1002/ana.410130302] [ Links ]
18. McDonald WI, Compston A, Edan G, et al. Recommended diagnostic criteria for multiple sclerosis: Guidelines from the international panel on the diagnosis of multiple sclerosis. Ann Neurol 2001;50(1):121-127. [http://dx.doi.org/10.1002/ana.1032] [ Links ]
19. Polman C, Reingold S, Edan G, et al. Diagnostic criteria for multiple sclerosis: 2005 revision to the 'McDonald Criteria. Ann Neurol 2005;58(6):840-846. [http://dx.doi.org/10.1002/ana.20703] [ Links ]
20. Polman CH, Reingold SC, Banwell B, et al. Diagnostic criteria for multiple sclerosis: 2010 Revisions to the McDonald Criteria. Ann Neurol 2011;69(2):292-302. [http://dx.doi.org/10.1002/ana.22366] [ Links ]
21. Swanton JK, Rovira A, Tintoré M, et al. MRI criteria for multiple sclerosis in patients presenting with clinically isolated syndromes: A multicentre retrospective study. Lancet Neurol 2007;6(8):677-686. [http://dx.doi.org/10.1016/S1474-4422(07)70176-X] [ Links ]
22. Rovira A, Swanton JK, Tintoré M, et al. A single, early magnetic resonance imaging study in the diagnosis of multiple sclerosis. Arch Neurol 2009;66(5):587-592. [http://dx.doi.org/10.1001/ archneuroL2009.49] [ Links ]
23. Montalban X, Tintoré M, Swanton JK, et al. MRI criteria for MS in patients with clinically isolated syndromes. Neurology 2010;74(5):427-434. [http://dx.doi.org/10.1212/WNL.0b013e3181cec45c1 [ Links ]
24. Miller DH, Weinshenker BG, Filippi M, et al. Differential diagnosis of suspected multiple sclerosis: A consensus approach. Mult Scler 2008;14(9):1157-1174. [http://dx.doi. org/10.1177/1352458508096878] [ Links ]
25. Lennon VA, Wingerchuk DM, Kryzer TJ, et al. A serum antibody marker for neuromyelitis optica:Distinction from multiple sclerosis. Lancet 2004;364(9451):2106-2112. [http://dx.doi.org/10.1016/S0140-6736(04)17551-X] [ Links ]
26. Wingerchuk DM, Lennon VA, Pittock SJ, et al. Revised diagnostic criteria for neuromyelitis optica. Neurology 2006;66(10):1485-1489. [http://dx.doi.org/10.1212/01.wnl.0000216139.44259.74] [ Links ]
27. Freedman MS, Patry DG, Grand'Maison F, et al. Treatment optimization in multiple sclerosis. Can J Neurol Sci 2004;31(2):157-168. [ Links ]
28. O'Connor P, Devonshire V for the Canadian Network of MS Clinics. The use of disease-modifying agents in multiple sclerosis - by the Canadian Network of MS Clinics. Can J Neurol Sci 2008; 35:127-129. [ Links ]
29. Gold R, Montalban X. Multiple sclerosis: More pieces of the immunological puzzle. Lancet Neurol 2012;11(1):9-10. [http://dx.doi.org/10.1016/S1474-4422(11)70268-X] [ Links ]
30. Comi G, Filippi M, Barkhof F, et al. Effect of early interferon treatment on conversion to definite multiple sclerosis: A randomised study. Lancet 2001;357(9268):1576-1582. [http://dx.doi.org/10.1016/S0140-6736(00)04725-5] [ Links ]
31. Comi G, De Stefano N, Freedman M, et al. Comparison of two dosing frequencies of subcutaneous interferon beta-1a in patients with a first clinical demyelinating event suggestive of multiple sclerosis (REFLEX): A phase 3 randomised controlled trial. Lancet Neurol 2012;11(1):33-41. [http://dx.doi.org/10.1016/S1474-4422(11)70262-9] [ Links ]
32. Kinkel RP, Kollman C, O'Connor P, et al. IM interferon beta-1a delays definite multiple sclerosis 5 years after a first demyelinating event. Neurology 2006;66(5):678-684. [http://dx.doi.org/10.1212/01.wnl.0000200778.65597.ae1 [ Links ]
33. Kappos L, Freedman MS, Polman CH, et al. Long-term effect of early treatment with interferon beta-1b after a first clinical event suggestive of multiple sclerosis: 5-year active treatment extension of the phase 3 BENEFIT trial. Lancet Neurol 2009;8(11):987-997. [http://dx.doi.org/10.1016/S1474-4422(09)70237-6] [ Links ]
34. Kappos L, Weinshenker B, Pozzilli C, et al. Interferon beta-1b in secondary progressive MS: A combined analysis of the two trials. Neurology 2004;63(10):1779-1787. [http://dx.doi.org/10.1212/01.WNL.0000145561.08973.4F] [ Links ]
35. Cohen JA, Antel JP. Does interferon beta help in secondary progressive MS? Neurology 2004;63(10):1768-1769. [http://dx.doi.org/10.1212/01.WNL.0000147171.32178.481] [ Links ]
36. Association of British Neurologists. ABN guidelines for treatment of multiple sclerosis with beta-interferon and glatiramer acetate 2007. London: Association of British Neurologists, 2007. [ Links ]
37. Goodin DS, Frohman EM, Garmany JR, et al. Disease modifying therapies in multiple sclerosis: Subcommittee of the American Academy of Neurology and the MS Council for Clinical Practice Guidelines. Neurology 2002;58(2):169-178. [http://dx.doi.org/10.1212/WNL.58.2.1691 [ Links ]
38. Rojas JI, Romano M, Ciapponi A, Patrucco L, Cristiano E. Interferon beta for primary progressive multiple sclerosis. Cochrane Database of Systematic Reviews 2009;1:CD006643. [http://dx.doi.org/10.1002/14651858.CD006643.pub21 [ Links ]
39. Barnes D, Hughes RA, Morris RW, et al. Randomised trial of oral and intravenous methylprednisolone in acute relapses of multiple sclerosis. Lancet 1997;349(9056):902-906. [http://dx.doi.org/10.1016%2FS0140-6736%2896%2906453-71 [ Links ]
40. Markowitz CE. Interferon-beta: Mechanism of action and dosing issues. Neurology 2007;68(24 Suppl 4):S8-S11; [http://dx.doi.org/10.1212%2F01.wnl.0000277703.74115.d21 [ Links ]
41. Weber MS, Hohlfeld R, Zamvil SS. Mechanism of action of glatiramer acetate in treatment of multiple sclerosis. Neurotherapeutics 2007;4(4):647-653. [http://dx.doi.org/10.1016%2Fj.nurt.2007.08.0021 [ Links ]
42. The IFNB Multiple Sclerosis Study Group. Interferon beta-1b is effective in relapsing-remitting multiple sclerosis. Clinical results of a multicenter, randomised, double-blind, placebo-controlled trial. Neurology 1993;43(4):655-661. [http://dx.doi.org/10.1016/S0140-6736(98)10039-9] [ Links ]
43. Jacobs LD, Cookfair DL, Rudick RA, et al. Intramuscular interferon beta-1a for disease progression in relapsing multiple sclerosis. Ann Neurol 1996;39(3):285-294. [http://dx.doi.org/10.1002/ana.410390304] [ Links ]
44. PRISMS (Prevention of Relapses and Disability by Interferon beta-1a Subcutaneously in multiple sclerosis) Study Group. Randomised double-blind placebo-controlled study of interferon beta-1a in relapsing/remitting multiple sclerosis. Lancet 1998;7:352(9139):1498-1504. [http://dx.doi.org/10.1016%2FS0140-6736%2898%2903334-0] [ Links ]
45. Johnson KP, Brooks BR, Cohen JA, et al. Copolymer 1 reduces relapse rate and improves disability in relapsing-remitting multiple sclerosis: Results of a phase III multicenter, double-blind placebo- controlled trial. Neurology 1995;45(7):1268-1276. [http://dx.doi.org/10.1212%2FWNL.45.7.1268] [ Links ]
46. Jacobs LD, Beck RW, Simon JH, et al. Intramuscular interferon beta-1a therapy initiated during a first demyelinating event in multiple sclerosis. N Engl J Med 2000;343(13):898-904. [http://dx.doi.org/10.1056/NEJM2000092834313011. [ Links ]
47. Kappos L, Polman CH, Freedman MS, et al. Treatment with interferon beta-1b delays conversion to clinically definite and McDonald MS in patients with clinically isolated syndromes. Neurology 10;67(7):1242-1249. [http://dx.doi.org/10.1212%2F01.wnl.0000237641.33768.8d1 [ Links ]
48. Comi G, Martinelli V, Rodegher M, et al; PreCISe study group. Effect of glatiramer acetate on conversion to clinically definite multiple sclerosis in patients with clinically isolated syndrome (PreCISe study): A randomised, double-blind, placebo-controlled trial. Lancet 2009;374(9700):1503-1511. [http://dx.doi.org/10.1016%2FS0140-6736%2809%2961259-91 [ Links ]
49. Kappos L; European Study Group on interferon beta-1b in secondary progressive MS Placebo-controlled multicentre randomised trial of interferon beta-1b in treatment of secondary progressive multiple sclerosis. Lancet 1998;352(9139):1491-1497. [http://dx.doi.org/10.1016/S0140-6736(98)10039-9] [ Links ]
50. Secondary Progressive Efficacy Clinical Trial of Recombinant Interferon-Beta-1a in MS (SPECTRIMS) Study Group. Randomised controlled trial of interferon- beta-1a in secondary progressive MS. Neurology 2001;56(11):1496-1504. [http://dx.doi.org/10.1212/WNL.56.11.14961 [ Links ]
51. Mikol DD, Barkhof F, Chang P, et al; REGARD study group. Comparison of subcutaneous interferon beta-1a with glatiramer acetate in patients with relapsing multiple sclerosis (the REbif v. Glatiramer Acetate in Relapsing MS Disease [REGARD1 study): A multicentre, randomised, parallel, open-label trial. Lancet Neurol 2008;7(10):903-914. [http://dx.doi.org/10.1016/S1474-4422(08)70200-X1 [ Links ]
52. O'Connor P, Filippi M, Arnason B, et al. 250 µg or 500 µg interferon beta-1b versus 20 mg glatiramer acetate in relapsing-remitting multiple sclerosis: A prospective, randomised, multicenter study. Lancet Neurol 2009;8(10):889-897. [http://dx.doi.org/10.1016/S1474-4422(09)70226-1] [ Links ]
53. Durelli L, Verdun E, Barbero P, et al; Independent Comparison of Interferon (INCOMIN) Trial Study Group. Every-other-day interferon beta-1b versus once-weekly interferon beta-1a for multiple sclerosis: results of a 2-year prospective randomised multicentre study (INCOMIN). Lancet 2002;359(9316):1453-1460. [http://dx.doi.org/10.1016/S0140-6736(02)08430-1] [ Links ]
54. Panitch H, Goodin DS, Francis G, et al; EVIDENCE (EVidence of Interferon Dose-response: European North American Comparative Efficacy) Study Group and the University of British Columbia MS/MRI Research Group. Randomised, comparative study of interferon beta-1a treatment regimens in MS: The EVIDENCE Trial. Neurology 2002;59(10):1496-1506. [http:/dx.doi.org/10.1212/01.WNL.0000034080.43681.DA1 [ Links ]
55. Clanet M, Radue EW, Kappos L, et al; European IFNbeta-1a (Avonex) Dose-Comparison Study Investigators. A randomised, double-blind, dose-comparison study of weekly interferon beta-1a in relapsing MS. Neurology 2002;59(10):1507-1517. [http://dx.doi.org/10.1212/01. WNL.0000032256.35561.D6] [ Links ]
56. Kinkel RP, Dontchev M, Kollman C, Skaramagas TT, O'Connor PW, Simon JH; for the Controlled High-Risk Avonex multiple sclerosis Prevention Study in Ongoing Neurological Surveillance Investigators. Association between immediate initiation of intramuscular interferon beta-1a at the time of a clinically isolated syndrome and long-term outcomes: A 10-year follow-up of the controlled high-risk avonex multiple sclerosis prevention study in ongoing neurological surveillance. Arch Neurol 2012;69(2):183-190. [http://dx.doi.org/10.1001%2Farchneurol.2011.1426] [ Links ]
57. Goodin DS, Reder AT, Ebers GC, et al. Survival in MS: A randomized cohort study 21 years after the start of the pivotal IFNp-1b trial. Neurology 2012;78(17):1315-1322. [http://dx.doi.org/10.1212%2FWNL.0b013e3182535cf6] [ Links ]
58. Caon C, Din M, Ching W, Tselis A, Lisak R, Khan O. Clinical course after change of immunomodulating therapy in relapsing-remitting multiple sclerosis. Eur J Neurol 2006;13(5):471-474. [http://dx.doi.org/10.1111%2Fj.1468-1331.2006.01273.x] [ Links ]
59. Gajofatto A, Bacchetti P, Grimes B, High A, Waubant E. Switching first-line disease-modifying therapy after failure: Impact on the course of relapsing-remitting multiple sclerosis. Mult Scler 2009;15(1):50-58. [http://dx.doi.org/10.1177%2F13524585080966871 [ Links ]
60. Zwibel HL; Copolymer-1 Treatment Study Principal Investigators. Glatiramer acetate in treatment-naïve and prior interferon-beta-1b-treated multiple sclerosis patients. Acta Neurol Scand2006;113(6):378-386. [http://dx.doi.org/10.1111%2Fj.1600-0404.2006.00627.x1 [ Links ]
61. Carrá A, Onaha P, Luetic G, et al. Therapeutic outcome 3 years after switching of immunomodulatory therapies in patients with relapsing-remitting multiple sclerosis in Argentina. Eur J Neurol 2008;15(4):386-393. [http://dx.doi.org/10.1111%2Fj.1468-1331.2008.02071.x1 [ Links ]
62. EMA. Summary ofproduct characteristics: Gilenya. http://www.ema.europa.eu/docs/en_GB/document_ library/EPAR_-_Product_Information/human/002202/WC500104528.pdf (accessed 14 July 2013). [ Links ]
63. EMA. Summary of product characteristics: Tysabri. http://www.ema.europa.eu/docs/en_GB/ document_library/EPAR_-_Product_Information/human/000603/WC500044686.pdf (accessed 14 July 2013). [ Links ]
64. Marriott JJ, Miyasaki JM, Gronseth G, O'Connor PW; Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Evidence Report: The efficacy and safety of mitoxantrone (Novantrone) in the treatment of multiple sclerosis. Report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Neurology 2010;74(18):1463-1470. [http://dx.doi.org/10.1212%2FWNL.0b013e3181dc1ae0] [ Links ]
65. Hartung HP, Gonsette R, Konig N, et al. A placebo-controlled, double-blind, randomised, multicentre trial of mitoxantrone in progressive multiple sclerosis. Lancet 2002;360:2018-2025. [http://dx.doi.org/10.1016%2FS0140-6736%2802%2912023-X] [ Links ]
66. Millefiorini E, Gasperini C, Pozzilli C, et al. Randomized placebo-controlled trial of mitoxantrone in relapsing-remitting multiple sclerosis: 24-month clinical and MRI outcome. J Neurol 1997;244(3):153-159. [http://dx.doi.org/10.1007/s0041500500661 [ Links ]
67. Edan G, Miller D, Clanet M, et al. Therapeutic effect ofmitoxantrone combined with methylprednisolone in multiple sclerosis: A randomised multicentre study of active disease using MRI and clinical criteria. J Neurol Neurosurg Psychiatry 1997;62:112-118. [http://dx.doi.org/10.1136%2Fjnnp.62.2.112] [ Links ]
68. Le Page E, Leray E, Taurin G, et al. Mitoxantrone as induction treatment in aggressive relapsing remitting multiple sclerosis: Treatment response factors in a 5 year follow-up observational study of 100 consecutive patients. J Neurol Neurosurg Psychiatry 2008;79(1):52-56. [http://dx.doi.org/10.1136%2Fjnnp.2007.124958] [ Links ]
69. Ramtahal J, Jacob A, Das K, Boggild M. Sequential maintenance treatment with glatiramer acetate after mitoxantrone is safe and can limit exposure to immunosuppression in very active, relapsing remitting multiple sclerosis. J Neurol 2006;253(9):1160-1164. [http://dx.doi.org/10.1007%2Fs00415-006-0178-z] [ Links ]
70. Edan G, Comi G, Le Page E, Leray E, Rocca MA, Filippi M; French-Italian Mitoxantrone Interferon-beta-1b Trial Group. Mitoxantrone prior to interferon beta-1b in aggressive relapsing multiple sclerosis: A 3-year randomised trial. J Neurol Neurosurg Psychiatry 2011;82(12):1344-1350. [http:// dx.doi.org/10.1136%2Fjnnp.2010.229724] [ Links ]
71. Martinelli V, Amato MP, Bellantonio P. Incidence of acute leukaemia in multiple sclerosis patients treated with mitoxantrone. Neurol Sci Suppl 2008;29:S75. [http://dx.doi.org/10.1016%2FS0022-510X%2809%2970476-9] [ Links ]
72. Martinelli V, Radaelli M, Straffi L, Rodegher M, Comi G. Mitoxantrone: Benefits and risks in multiple sclerosis patients. Neurol Sci 2009;30(2):S167-S170. [http://dx.doi.org/10.1007%2Fs10072-009-0142-71 [ Links ]
73. Polman CH, O'Connor PW, Havrdova E, et al; AFFIRM Investigators. A randomised, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N Engl J Med 2006;354(9):899-910. [http://dx.doi.org/10.1056%2FNEJMoa0443971 [ Links ]
74. Rudick RA, Stuart WH, Calabresi PA, et al. Natalizumab plus interferon beta-1 for relapsing multiple sclerosis. N Engl J Med 2006;354:911-923. [http://dx.doi.org/10.1056%2FNEJMoa044396] [ Links ]
75. Goodman AD, Rossman H, Bar-Or A, et al GLANCE. Results ofa phase II, randomised, double-blind, placebo- controlled study. Neurology 2009;72:806-812. [http://dx.doi.org/10.1212/01.wnl.0000343880.13764.691 [ Links ]
76. Kappos L, Bates D, Edan G, et al. Natalizumab treatment for multiple sclerosis: Updated recommendations for patient selection and monitoring. Lancet Neurol 2011;10(8):745-758. [http:// dx.doi.org/10.1016%2FS1474-4422%2811%2970149-1] [ Links ]
77. Gorelik L, Bixler S, Cheung A, et al. Assessment of the incidence of anti-JCV antibodies in a cohort of natalizumab-treated patients with multiple sclerosis. Mult Scler 2010;16:S306-S307. [ Links ]
78. Trampe AK, Hemmelmann C, Stroet A, et al. Anti-JC virus antibodies in a large German natalizumab-treated multiple sclerosis cohort. Neurology 2012;78(22):1736-1742. [http://dx.doi.org/10.1212%2FWNL.0b013e3182583022] [ Links ]
79. O'Connor PW, Goodman A, Kappos L, et al. Disease activity return during natalizumab treatment interruption in patients with multiple sclerosis. Neurology 2011;76(22):1858-1865. [http://dx.doi.org/10.1212%2FWNL.0b013e31821e7c8a]
80. Kappos L, Radue EW, O'Connor P, et al; FREEDOMS Study Group. A placebo-controlled trial of oral fingolimod in relapsing multiple sclerosis. N Engl J Med 2010;362(5):387-401. [http://dx.doi.org/10.1056%2FNEJMoa0909494] [ Links ]
81. Cohen JA, Barkhof F, Comi G, et al; TRANSFORMS Study Group. Oral fingolimod or intramuscular interferon for relapsing multiple sclerosis. N Engl J Med 2010;362(5):402-415. [http://dx.doi. org/10.1056%2FNEJMoa0907839] [ Links ]
82. Pelletier D, Hafler DA. Fingolimod for multiple sclerosis. N Engl J Med 2012;366(4):339-347. FREEDOMS AND TRANSFORMS [http://dx.doi.org/10.1056%2FNEJMct1101691] [ Links ]
83. Ben-Zacharia A, Lublin FD. Talking with your MS patients about difficult topics: Talking about initiating and adhering to treatment with injectable disease modifying agents. New York: National Multiple Sclerosis Society, 2009. [ Links ]
84. Carrá A, Macías-Islas AM, Gabbai AA, et al. Optimizing outcomes in multiple sclerosis: Consensus guidelines for the diagnosis and treatment of multiple sclerosis in Latin America. Ther Adv Neurol Disord 2011; 4(6):349-360. [http://dx.doi.org/10.1177%2F1756285611423560] [ Links ]
85. Arnold DL. Strategies for Monitoring and Assessing Therapy. Adv Stud Med 2006;6(7D):S701-S706. [ Links ]
86. Hirst C, Ingram G, Swingler R, Compston DA, Pickersgill T, Robertson NP. Change in disability in patients with multiple sclerosis: A 20-year prospective population-based analysis. J Neurol Neurosurg Psychiatry 2008;79:1137-1143. [http://dx.doi.org/10.1136%2Fjnnp.2007.133785] [ Links ]
87. Kurtzke JF. Rating neurologic impairment in multiple sclerosis. An expanded disability status scale (EDSS). Neurology 1983;33(11):1444-1452. [http://dx.doi.org/10.1212%2FWNL.33.11.1444] [ Links ]
88. Ransohoff R. A fundamentally new view of multiple sclerosis. Int J MS Care 2000;2(2):2-8. [htttp:// dx.doi.org/10.7224%2F1537-2073-2.2.2] [ Links ]
89. Rovira A, Tintoré M, Alvarez-Cermeno JC, Izquierdo G, Prieto JM. Recommendations for using and interpreting magnetic resonance imaging in multiple sclerosis. Neurologia 2010;25:248-265. [http:// dx.doi.org/10.1016%2FS2173-5808%2810%2970049-3] [ Links ]
90. Butzkueven H, Kolbe SC, Jolley DJ, et al. Validation of linear cerebral atrophy markers in multiple sclerosis. J Clin Neurosci 2008 Feb;15(2):130-137. [http://dx.doi.org/10.1016%2Fj.jocn.2007.02.089] [ Links ]
91. Cohen BA, Khan O, Jeffery DR. Identifying and treating patients with suboptimal responses. Neurology 2004;63(Suppl 6):S33-S40 [http://dx.doi.org/10.1212%2FWNL.63.12_suppl_6.S33] [ Links ]
92. International Working Group for Treatment Optimization in MS. Treatment optimization in multiple sclerosis: Report of an international consensus meeting. Eur J Neurol 2004;11(1):43-47. [http://dx.doi.org/10.1046/j.1351-5101.2003.00711.x] [ Links ]
93. Hauser SL, Waubant E, Arnold DL, et al; HERMES Trial Group. B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. N Engl J Med 2008;358:676-688. [http://dx.doi.org/10.1056%2FNEJMoa0706383] [ Links ]
94. CAMMS223 Trial Investigators, Coles AJ, Compston DA, et al. Alemtuzumab vs. interferon beta-1a in early multiple sclerosis. N Engl J Med 359(17):1786-1801. [http://dx.doi.org/10.1056/ NEJMoa0802670] [ Links ]
95. Krishnan C, Kaplin AI, Brodsky RA, et al. Reduction of disease activity and disability with high-dose cyclophosphamide in patients with aggressive multiple sclerosis. Arch Neurol 2008;65(8):1044-1051. [http://dx.doi.org/10.1001%2Farchneurol.65.8.noc80042] [ Links ]
96. Coles AJ, Cox A, Le Page E, et al. The window of therapeutic opportunity in multiple sclerosis. J Neurol 2006;253(1):98-108. [http://dx.doi.org/10.1007%2Fs00415-005-0934-5] [ Links ]
97. Kappos L, Calabresi P, O'Connor P, et al. Efficacy and safety of ocrelizumab in patients with relapsing-remitting multiple sclerosis: Results of a phase II randomised placebo-controlled multicentre trial. Mult Scler 2010;16(Suppl 7):S33. [ Links ]
98. Bar-Or A, Calabresi PA, Arnold D, et al. Rituximab in relapsing-remitting multiple sclerosis: A 72-week, open-label, phase I trial. Ann Neurol 2008;63(3):395-400. [http://dx.doi.org/10.1002%2Fana.21363] [ Links ]
99. Bielekova B, Richert N, Howard T, et al. Humanized anti-CD25 (daclizumab) inhibits disease activity in multiple sclerosis patients failing to respond to interferon beta. Proc Natl Acad Sci U S A 2004;101(23):8705-8708 [http://dx.doi.org/10.1073%2Fpnas.0402653101] [ Links ]
100. Rose JW, Burns JB, Bjorklund J, et al. Daclizumab phase II trial in relapsing and remitting multiple sclerosis: MRI and clinical results. Neurology 2007;69(8):785-789. [http://dx.doi.org/10.1212%2F01. wnl.0000267662.41734.1f] [ Links ]
101. Wynn D, Kaufman M, Montalban X, et al. Daclizumab in active relapsing multiple sclerosis (CHOICE study): A phase 2, randomised, double-blind, placebo-controlled, add-on trial with interferon beta. Lancet Neurol 2010;9(4):381-390. [http://dx.doi.org/10.1016%2FS1474-4422%2810%2970033-8] [ Links ]
102. Kappos L, Gold R, Miller DH, et al. Efficacy and safety of oral iumarate in patients with relapsing-remitting multiple sclerosis: A multicentre, randomised, double- blind, placebo-controlled phase IIb study. Lancet 2008;372(9648):1463-1472. [http://dx.doi.org/10.1016%2FS0140-6736%2808%2961619-0] [ Links ]
103. Comi G, Pulizzi A, Rovaris M, et al. Effect of laquinimod on MRI- monitored disease activity in patients with relapsing-remitting multiple sclerosis: A multicentre, randomised, double-blind, placebo-controlled phase IIb study. Lancet 2008;371(9630):2085-2892. [http://dx.doi.org/10.1016%2 FS0140-6736%2808%2960918-6] [ Links ]
104. Comi G, Abramsky O, Arbizu T, et al. Oral laquinimod in patients with relapsing-remitting multiple sclerosis: 36-week double-blind active extension of the multi-centre, randomized, double- blind, parallel-group placebo-controlled study. Mult Scler 2010;16(11):1360-1366. [http://dx.doi. org/10.1177%2F1352458510378127] [ Links ]
105. Confavreux C, Li DK, Freedman MS, et al; Teriflunomide Multiple Sclerosis Trial Group. Long-term follow-up of a phase 2 study of oral teriflunomide in relapsing multiple sclerosis: Safety and efficacy results up to 8.5 years. Mult Scler 2012;18(9):1278-1289. [http://dx.doi.org/10.1177/1352458512436594] [ Links ]
106. O'Connor P, Wolinsky JS, Confavreux C, et al. A placebo-controlled phase III trial (TEMSO) of oral teriflunomide in relapsing multiple sclerosis: Clinical efficacy and safety outcomes. Mult Scler 2010;16:S23. [ Links ]
107. Krupp LB, Coyle PK, Doscher C, et al. Fatigue therapy in multiple sclerosis: Results of a double-blind, randomised, parallel trial of omantidine, pemoline and placebo. Neurology 1995;45(11):1956-1961. [http://dx.doi.org/10.1212/WNL.45.11.1956] [ Links ]
108. Krupp LB, Christodoulou C, Melville P, et al. Donepezil improved memory in multiple sclerosis in a randomised clinical trial. Neurology 2004;63(9):1579-1585. [http://dx.doi.org/10.1212/01. WNL.0000142989.09633.5A] [ Links ]
109. Lovera JF, Frohman E, Brown TR, et al. Memantine for cognitive impairment in multiple sclerosis: A randomised placebo-controlled trial. Mult Scler 2010;16(6):715-723. [http://dx.doi.org/10.1177%2F1352458510367662] [ Links ]
110. Boisy AR, Cohen JA. Multiple sclerosis symptom management. Expert Rev Neurother 2007;7(9):1213-1222. [http://dx.doi.org/10.1586%2F14737175.7.9.1213] [ Links ]
111. Hyman N, Barnes M, Bhakta B, et al. Botulinum toxin (Dysport®) treatment of hip adductor spasticity in multiple sclerosis: a prospective, randomised, double blind, placebo controlled, dose ranging study. J Neurol Neurosurg Psychiatry 2000;68:707-712. [http://dx.doi.org/10.1136%2Fjnnp.68.6.707] [ Links ]
112. Feldman RG, Kelly-Hayes M, Conony JP, et al. Baclofen for spasticity in multiple sclerosis: Double-blind crossover and three-year study. Neurology 1978;28(11):1094. [http://dx.doi.org/10.1212%2FWNL.28.11.1094] [ Links ]
113. Mueller ME, Gruenthal M, Olsen WL, et al. Gabapentin for relief of upper motor neuron symptoms in multiple sclerosis. Arch Phys Med Rehabil 1997;78:521-524. [ Links ]
114. Fowler CJ, Panicker JN, Drake M. A UK consensus on the management of the bladder in multiple sclerosis. Postgrad Med J 2009;85:552-559. [http://dx.doi.org/10.1136%2Fjnnp.2008.159178] [ Links ]
115. Kalsi V, Gonzales G, Popat R, et al Botulinum injections for the treatment of bladder symptoms of multiple sclerosis. Ann Neurol 2007;62(5):452-457. [http://dx.doi.org/10.1002%2Fana.21209] [ Links ]
116. Safarinejad, MR. Evaluation of the safety and efficacy of sildenafil citrate for erectile dysfunction in men with multiple sclerosis: A double-blind, placebo controlled, randomized study. J Urol 2009;181(1):252-258. [http://dx.doi.org/10.1016%2Fj.juro.2008.09.003] [ Links ]
117. Cruccu G, Gronseth G, Alksne J, et al. AAN-EFNS guidelines on trigeminal neuralgia management. Eur J Neurol 2008;15(10):1013-1028. [http://dx.doi.org/10.1111%2Fj.1468-1331.2008.02185.x] [ Links ]
118. Koller, WC. Pharmacologic trials in the treatment of cerebellar tremor. Arch Neurol 1984;41(3):280-281. [http://dx.doi.org/10.1001%2Farchneur.1984.04050150058017] [ Links ]
119. Goodman AD, Brown TR, Cohen JA, et al. Dose comparison trial of sustained-release fampridine in multiple sclerosis. Neurology 2008;71:1134-1141. [http://dx.doi.org/10.1212%2F01.wnl.0000326213.89576.0e] [ Links ]
120. Ashburn A, De Souza L. An approach to the management of multiple sclerosis. Physiother Theory Pract 1988;4(3):139-145. [http://dx.doi.org/10.3109/09593988809159064] [ Links ]
121. Rietberg M, Brooks D, Uitdehaag B, Kwakkel G. Exercise therapy for multiple sclerosis. Cochrane Library 2003. [http://dx.doi.org/10.1002%2F14651858.CD003980] [ Links ]
122. Thompson AJ. Symptomatic management and rehabilitation in multiple sclerosis. J Neurol Neurosurg Psychiatry 2001;71:ii22-ii27. [http://dx.doi.org/10.1136/jnnp.71.suppl_2.ii22] [ Links ]
123. Wiles RG, Newcombe KJ, Fuller S, et al Controlled randomised crossover trial of the effects of physiotherapy on mobility in chronic multiple sclerosis. J Neurol Neurosurg Psychiatry 2001;70:174-179. [http://dx.doi.org/10.1136%2Fjnnp.70.2.174] [ Links ]
124. Zifko UA. Management of fatigue in patients with multiple sclerosis. Drugs 2004;64(12):1295-1304. [http://dx.doi.org/10.2165%2F00003495-200464120-00003] [ Links ]
125. Kielhofner G. Model of Human Occupation. 4th ed. Philadelphia: Lippincott Williams and Wilkins, 2007. [ Links ]
126. Law, M. Enabling Occupation: An Occupational Therapy Perspective. In: Townsend E, ed. Ottawa: Canadian Association of Occupational Therapists, 2002: Chapters 1 and 3. [ Links ]
127. National Institute for Clinical Excellence. Multiple Sclerosis: Management of Multiple Sclerosis in Primary And Secondary Care. London: NICE, 2004. [ Links ]
128. Kraft G, Freal J, Coryell J. Disability, disease duration, and rehabilitation service needs in multiple sclerosis: patient perspectives. Arch Phys Med Rehabil 1986;67(3):164-168. [http://dx.doi.org/10.1016/0003-9993(86)90060-2] [ Links ]
129. Mann W, Ottenbacher K, Fraas L. Effectiveness of assistive technology and environmental interventions in maintaining independence and reducing home care costs for the elderly. Arch Fam Med 1999;8:210-217. [ Links ]
130. Harrison S. Fatigue Management for People with Multiple Sclerosis. 2nd ed. London: College of Occupational Therapists, 2007. [ Links ]
131. Pedrettti LW, Zoltan B. Occupational Therapy Skills for Physical Dysfunction. 3rd ed. St Louis: Mosby, 1990. [ Links ]
132. Bowcher H, May M. Occupational therapy for the management of fatigue in multiple sclerosis. Br J Occup Ther 1998;61(11):488-492. [ Links ]
133. Shapiro R. Symptom management in multiple sclerosis. Ann Neuro 1994;36(S1):S123-S129. [htttp://dx.doi.org/10.1002%2Fana.410360730] [ Links ]
134. Hubsky E, Sears J. Fatigue in multiple sclerosis: Guidelines for nursing care. Rehabil Nurs 1992;17(4):176-180. [http://dx.doi.org/10.1002%2Fj.2048-7940.1992.tb01542.x] [ Links ]
135. Rosenberg J, Shafor R. Fatigue in multiple sclerosis: A rational approach to evaluation and treatment. Curr Neurol Neurosci Rep 2005;5(2):140-146. [http://dx.doi.org/10.1007/s11910-005-0012-5] [ Links ]
136. Welham, L. Occupational therapy for fatigue in patients with multiple sclerosis. Br J Occup Ther 1995;58(12):507-509. [ Links ]
 Correspondence:
Correspondence:
D L Giampaolo
(docman1@me.com)


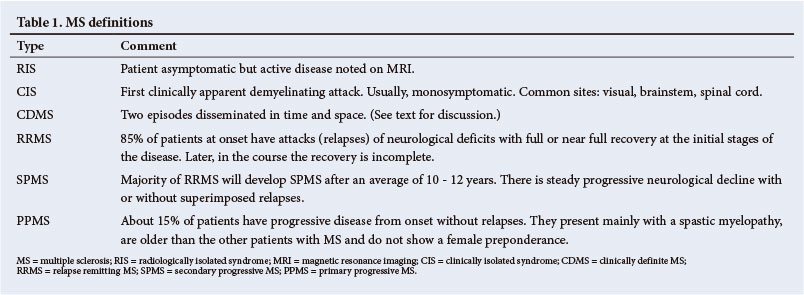{kind=link}
{kind=link}
{kind=link}
{kind=link}
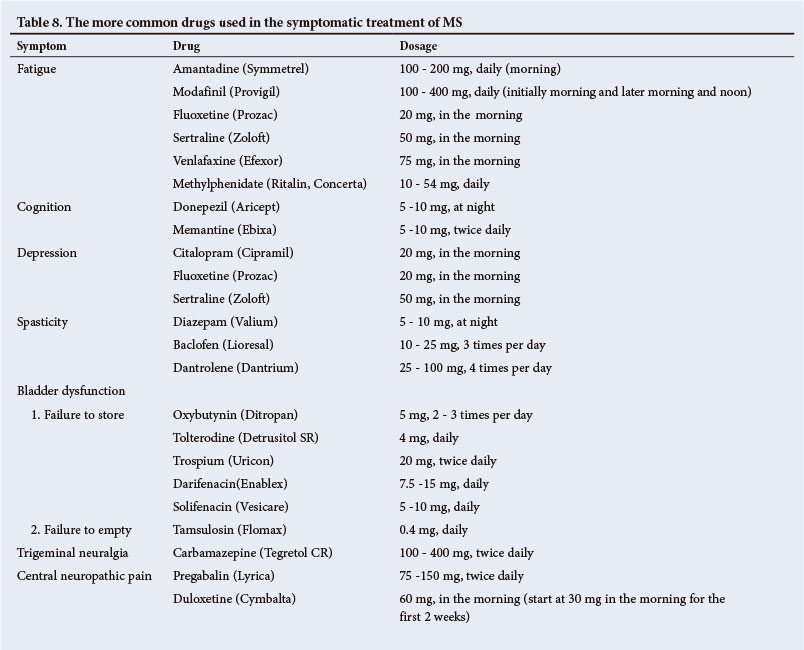{kind=link}
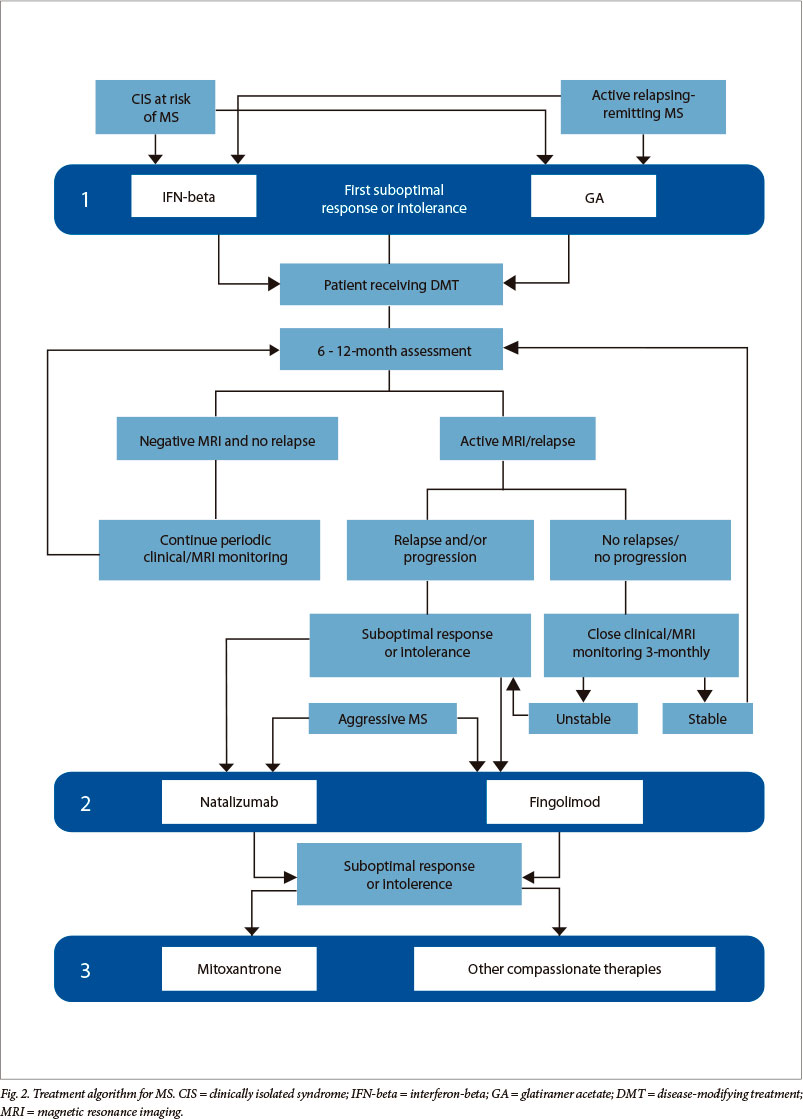{kind=link}