










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSAMJ: South African Medical Journal
On-line version ISSN 2078-5135
Print version ISSN 0256-9574
SAMJ, S. Afr. med. j. vol.103 n.5 Pretoria May. 2013
GUIDELINE
South African guideline for the management of chronic hepatitis B: 2013
C W N SpearmanI; M W SonderupXI, II; J F BothaXII, III; S W van der MerweIV, V ; E SongVI, VII; C KassianidesVIII, IX; K A NewtonXIII, X ; H N HairwadziXIV
IMB ChB, FCP (SA), MMed, PhD; Division of Hepatology, Department of Medicine, University of Cape Town, South Africa
IIMB ChB, BPharm, FCP (SA.) South African Gastroenterology Society, Mowbray, Cape Town, South Africa
IIIMB ChB, FCP (SA). Sandton Clinic, Bryanston, Johannesburg, South Africa
IVMB ChB, MSc, MMed, PhD. Department of Immunology, University of Pretoria, South Africa
VMB ChB, MSc, MMed, PhD. Department of Clinical and Experimental Medicine, University of Leuven, Flanders, Belgium
VIMB ChB, FCP (SA), FRCP (London). Department of Medicine, University of the Witwatersrand, Johannesburg, South Africa
VIIMB ChB, FCP (SA), FRCP (London). Donald Gordon Medical Centre, Johannesburg, South Africa
VIIIMB ChB, FCP (SA). Morningside Clinic, Sandton, Johannesburg, South Africa
IXMB ChB, FCP (SA). Gastroenterology Foundation of South Africa, Mowbray, Cape Town, South Africa
XMB ChB, FCP (SA)Department of Gastroenterology, Division of Clinical Medicine, University of KwaZulu-Natal, Durban, South Africa
XIMB ChB, BPharm, FCP (SA). Division of Hepatology, Department of Medicine, University of Cape Town, South Africa
XIIMB ChB, FCP (SA). South African Gastroenterology Society, Mowbray, Cape Town, South Africa
XIIIMB ChB, FCP (SA).South African Gastroenterology Society, Mowbray, Cape Town, South Africa
XIVMB ChB, MMed, PhD Division of Hepatology, Department of Medicine, University of Cape Town, South Africa
ABSTRACT
Hepatitis B remains a significant yet preventable health issue in South Africa. The introduction of the hepatitis B vaccine into the country some 18 years ago has demonstrated benefit, but the exposure to, and prevalence of chronic HBsAg positivity remain unacceptably high. Those with chronic hepatitis B virus infection have an elevated risk of developing cirrhosis with end-stage liver disease and a markedly elevated risk of hepatocellular carcinoma, independent of the presence of cirrhosis.
The challenge in South Africa remains prevention through the universal vaccination coverage of all children and the identification of those with chronic hepatitis B virus infection. Over the last decade our understanding of hepatitis B and its behaviour and natural history in those with chronic infection has significantly improved. This understanding is key to identifying those who warrant further evaluation and therapy. A number of global societies have updated their guidelines in recent years. This document draws on these guidelines and serves to contextualise, for South Africa, practice guidelines for the management of chronic hepatitis B.
1. Introduction
Hepatitis Β is an important public health issue in South Africa (SA). Prior to the introduction of the hepatitis B vaccine into the South African Expanded Programme of Immunisation (EPI) in 1995, prevalence rates of this disease were 0.3 - 15%.[1] However, unlike countries such as Taiwan,[2] SA has had no catch-up vaccination programme to ensure complete vaccination coverage. In addition, the HIV/AIDS pandemic has had a potentially deleterious influence on the natural history of patients co-infected with HIV and the hepatitis B virus (HBV).[3]
The spectrum of disease and natural history of chronic HBV infection is diverse, ranging from a low viraemic immune control state to progressive chronic hepatitis, with the potential for the ensuing complications of cirrhosis, liver failure and hepatocellular carcinoma (HCC).[4] As understanding of the natural history of chronic hepatitis B increased over the past decade, there have been significant therapeutic advances. The decision to treat and the choice of therapy is dependent on both the phase of chronic infection and patient factors.
This guideline draws on the recently published guidelines by the American Association for the Study of Liver Disease (AASLD), the European Association for the Study of the Liver (EASL), the Asia-Pacific Association for the Study of the Liver (APASL), National Institutes of Health (NIH) and the World Gastroenterology Organisation (WGO).[5-9] It serves as an attempt to contextualise practice guidelines on the management of chronic hepatitis B in SA.
2. Pathogenesis and natural history
See Table 1. Hepatitis is an enveloped partially double-stranded DNA virus belonging to the Hepadnaviridae family. It is 100 times more infectious than HIV and can be transmitted by perinatal, percutaneous and sexual exposure.[10] Close person-to-person contact is an important form of transmission, most notably among children in highly endemic areas, such as in SA.[5,101
Liver injury due to hepatitis B is mainly caused by cellular immune mediated mechanisms with cytotoxic T lymphocyte lysis of infected hepatocytes. The magnitude of the individual's adaptive cellular immune response to HBV-related antigens determines the outcome of acute HBV infection, as well as the degree of liver injury. Chronically infected patients are unable to sustain an immune response to HBV and may experience intermittent episodes of hepatocyte destruction in an attempt to clear virally infected hepatocytes, in what can be termed 'flares'. Note that, during the acute infection, hepatitis B does not appear to induce an intra-hepatic innate immune response. Instead, it acts as a 'stealth' virus early in the infection.[9]
Age is also an important host factor determining the risk of chronicity. Following acute exposure to HBV, 90% of neonates born to hepatitis B 'e' antigen (HBeAg)-positive mothers, 20 - 50% of infants and children under the age of 5 years, and <5% of adults will develop chronic hepatitis B infection.[11,121 Viral variants may also influence the course and outcome of the disease. In addition, and only rarely and in the setting of profound immune suppression, the virus can be directly cytopathic.
In choosing an appropriate management strategy, a clear understanding of the process of hepatitis B viral replication, as well as the natural history of chronic hepatitis B, is vital:
Following acute exposure, the HBV enters the hepatocyte and is imported into the nucleus. The partially doubled-stranded DNA is repaired to form a circular extra-chromosomal molecule called the covalently closed circular DNA (cccDNA),[131 which is the transcriptional template for the viral messenger RNAs (mRNAs). The RNA form of the genome is encapsidated together with the reverse transcriptase, and reverse transcription occurs within the cytoplasm. Cytoplasmic viral capsids containing mature viral DNA are either transported to the nucleus, thereby replenishing cccDNA, or bind to HBV surface antigens which have accumulated in the endoplasmic reticulum, bud through the cellular membranes and are secreted from the hepatocyte non-cytopathically, as virions.
Hence, even if the individual clears hepatitis B surface antigen (HBsAg), the hepatocyte still harbours cccDNA. This is the basis of occult HBV infection, which is defined as detectable HBV DNA in the liver and a very low level (<200 IU/ml) of HBV DNA in the blood of those previously exposed to HBV, viz. HBsAg negative and hepatitis B immunoglobulin G core antibody (anti-HBc IgG) positive. The clinical significance of occult HBV is that immunosuppression may lead to reactivation in these patients. HBV DNA can also integrate into the cellular genome during chronic infection, as a result of random insertion of viral DNA into the host genome, by host processes during failed repair of the partially double-stranded DNA. This integrated DNA plays no role in viral replication, but plays an important and ill-defined role in the development of HCC.
There are 5 phases of chronic infection which are not necessarily sequential and are of variable duration.'6,14
1. The immune tolerant phase is characterised by HBeAg posi-tivity, high levels of viral replication (high serum HBV DNA), normal transaminases, minimal or no hepatic necroinflammation and no or slow progression to fibrosis. During this phase, the rate of spontaneous HBeAg loss is low. This phase, which is more common and more prolonged in individuals infected perinatally or under the age of 5 years, frequently persists into early adulthood and is frequent in SA.
2. The immune clearance phase (HBeAg-positive chronic hepatitis B) is characterised by HBeAg positivity, but lower levels of viral replication. The transaminases are elevated and histologically there is more severe necroinflammation and more rapid progression of fibrosis. This phase may last several weeks to years and, if successful, a sustained HBeAg seroconversion will occur with the development of anti-HBe. A successful HBeAg seroconversion is more likely to occur in individuals infected during adulthood.
3. The inactive HBV carrier or latency state (immune control phase) follows successful HBeAg to anti-HBe seroconversion and is characterised by very low (<2 000 IU/ml) or undetectable HBV DNA levels and normal transaminases. As a result of immunological control of the infection, these patients have a good prognosis, with a much lower risk of progression to cirrhosis or HCC. HBsAg loss and seroconversion to anti-HBs may occur spontaneously at a rate of 1 - 3% per year.
4. Five to 15% of individuals in the inactive HBV carrier state will develop HBeAg-negative chronic hepatitis B. This reactivation phase represents a later phase in the natural history of the disease and is more common in older men. Nucleotide substitutions in the precore and/or basal core promoter regions of the HBV genome result in HBV variants that are unable to express HBeAg, or which do so at very low levels. This phase is characterised by HBeAg negativity, fluctuating transaminases and HBV DNA levels, significant necroinflammation and progressive fibrosis. Low levels of hepatitis B immunoglobulin M core antibody (anti-HBc IgM) may be detected.
It is important, but often difficult, to distinguish this phase from the inactive HBV carrier state. Patients with HBeAg-negative chronic hepatitis B have a high risk of progression to cirrhosis, which may in turn lead to decompensation and the risk of HCC. At least 1 year follow-up, with 3 - 4-monthly monitoring of alanine transaminase (ALT) and HBV DNA levels, is required to confidently distinguish these two phases of the disease.[15-17]
Individuals in the inactive HBV carrier state may also revert back to HBeAg positivity and develop HBeAg-positive disease.
5. Occult HBV infection is the term used to describe those cases where patients have cleared surface antigen but have detectable plasma HBV DNA. Serologically they are HBsAg negative, hepatitis B surface antibody (HBsAb) positive and anti-HBc IgG positive, yet they are positive for HBV DNA, albeit at very low levels (invariably <200 IU/ml). While no liver disease is associated with occult infection, these individuals are at very high risk of reactivation of HBV with immune suppression, e.g. during use of rituximab (MabTheraR), and require prophylactic antiviral therapy.
3. Diagnosis
In diagnosing chronic hepatitis B, HBV serological markers and HBV DNA levels must be carefully and correctly interpreted, to accurately decide on the phase of the chronic infection so that appropriate management, if required, can be instituted.
3.1 HBV serological markers[5,18]
HBsAg:
• General and screening marker of infection
• First serological marker to appear
• Surrogate marker for transcriptionally active cccDNA
• Infection is considered chronic if HBsAg persists for >6 months.
HBeAg:
• Indicates active replication of virus
• Absent or low in pre-core or basal core promoter mutations.
Anti-HBc total (HBcAb total):
• Includes both IgG and IgM HBcAb.
IgG anti-HBc:
• Most sensitive marker of past exposure to HBV as anti-HBs may be undetectable if HBV infection was acquired in childhood, as is common in SA.
IgM anti-HBc:
• Marker of acute infection or reactivation
• Strongly positive in acute infection and possible low positivity in reactivation or flare.'191
Anti-HBs (HBsAb)
• Recovery and/or immunity to HBV
• Detectable after immunity is conferred by HBV vaccination.
Anti-HBe (HBeAb)
• Usually indicates HBeAg to anti-HBe seroconversion and that the virus is no longer replicating
• Also present in HBeAg-negative chronic hepatitis, with active replication due to mutants.
3.2 Virological evaluation of HBV infection
• Serum HBV DNA quantification
• HBV genotype
• HBV resistance testing.
3.3 Role of HBV DNA testing[18]
• Can differentiate chronic HBeAg-negative disease from the inactive latency state (HBV DNA <2 000 IU/ml)
• Differentiates between occult hepatitis B (IgG anti-HBc positive, HBV DNA positive, but <200 IU/ml) and resolved infection (IgG anti-HBc positive, anti-HBs positive, HBV DNA negative)
• Changes in HBV DNA levels used to monitor response to therapy
• In patients adherent to therapy, increasing HBV DNA levels indicate the emergence of resistant variants
• HBV DNA levels correlate with disease progression.'20-231
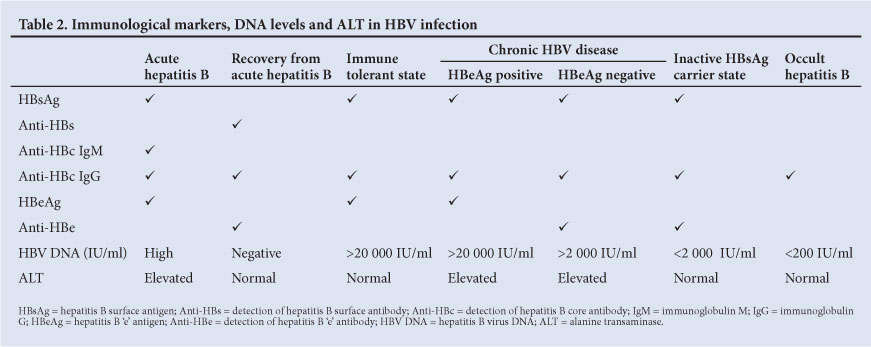
3.4 Immunological markers, DNA levels and ALT in HBV infection[16,24]
See Table 2.
4. Assessment of liver disease prior to therapy[5,6,16]
See Table 3.
4.1 Clinical history and physical examination
Include family history of HBV infection and HCC.
4.2 Assessment of the severity of the liver disease
• Liver profile including total bilirubin, conjugated bilirubin, ALT, aspartate transaminase (AST), alkaline phosphatase (ALP) and gamma-glutamyl transferase (GGT)
• Full blood count (FBC) including a differential count
• Serum albumin and international normalised ratio (INR) to assess synthetic function.
4.3 Viral serology
• HBsAg, anti-HBs, HBeAg and anti-HBe
• IgG anti-HBc (if assessing for occult HBV or previous cleared infection).
4.4 Viral replication
Serum HBV DNA quantified with real-time polymerase chain reaction (PCR).
4.5 Look for other co-factors
• Viral co-infection: HCV, HIV
• Non-alchoholic fatty liver disease/alcoholic liver disease
• Iron overload
• Drug/toxin-induced injury.
4.6 Liver biopsy
A liver biopsy is required to assess the degree of necroinflammation and fibrosis and is helpful in assessing the contribution of one or more comorbidities. It is generally indicated if the ALT is elevated and/or HBV DNA is >2 000 IU/ml, or when interferon-based therapy is being considered. A biopsy is useful in the SA context to assess the need for treatment, as there has often been a prolonged immune tolerant phase and liver enzymes may be only marginally elevated. A liver biopsy is not required in patients with clinical evidence of cirrhosis or when nucleos(t)ide analogue (NUC) therapy is indicated, regardless of the grade of activity or stage of fibrosis. The risk of severe complications with liver biopsy is low
(1/4 000 - 10 000).
4.7 ultrasound of the liver and Doppler studies of the portal vein
5. Goals and endpoints of therapy[5,6,16]
See Table 4. HBV infection cannot be eradicated completely with current available therapies because of the persistence of cccDNA, which acts as a viral reservoir in infected hepatocytes.'251 Even so, an ideal endpoint of treatment would be to achieve viral eradication with sustained HBsAg loss, with/without seroconversion to anti-HBs antibodies, as HBsAg is a surrogate marker for transcriptionally active cccDNA.'26,271 However, this is as yet uncommon and hence a broad goal of therapy is to prevent or reverse disease progression to cirrhosis, end-stage liver disease or HCC. This can be achieved by suppressing HBV replication, with a consequent improvement in necro-inflammation and fibrosis that lowers the risk of cirrhosis and HCC.'20-22,281

Once cirrhosis is established, preventing decompensation, HCC or death is the primary treatment goal. In those patients with early decompensation, suppression of HBV replication can improve synthetic function and decrease the Child-Pugh/ model for end-stage liver disease (MELD) score, and may delay the need for liver transplantation. In those with end-stage liver disease, suppression of HBV replication prior to transplantation reduces the risk of recurrence.
5.1 Endpoints of treatment: HBeAg-positive disease
• The ideal endpoint is sustained HBsAg loss due to therapy, with/without the development of anti-HBs
• Durable HBeAg loss and seroconversion to anti-HBe
• Durable suppression of HBV DNA to low or undetectable levels
• Normalisation of ALT.
5.2 Endpoints of treatment: HBeAg-negative disease
• The ideal endpoint is sustained HBsAg loss off therapy, with/ without the development of anti-HBs
• Durable suppression of HBV DNA to low or undetectable levels
• Normalisation of ALT.
6. Definitions of response[6]
Responses may be biochemical, serological, virological or histological and vary according to the type of therapy.
6.1 Interferon-based therapy
• Primary non-response is not well established
• A virological response is an HBV DNA concentration <2 000 IU/ml, evaluated at 6 months, end of therapy; and at 6 and 12 months after completion of therapy
• A sustained off-treatment virological response is defined as HBV DNA levels <2 000 IU/ml for at least 12 months after completion of treatment
• A serological response in patients with HBeAg-positive chronic hepatitis B is HBeAg seroconversion to anti-HBe positive.
6.2 NuC therapy
• Primary non-response is a <1 log10 IU/ml decrease in HBV DNA level from baseline at 3 months of therapy.
• A virological response is undetectable HBV DNA by real-time PCR assay within 48 weeks of therapy; evaluated every 3 - 6 months during therapy depending on the severity of the liver disease and type of NUC.
• Partial virological response is a >1 log10 IU/ml decrease in HBV DNA, but detectable HBV DNA by real-time PCR assay.
• A partial virological response should be assessed at 24 weeks for patients on lamivudine which is moderately potent but has a low genetic barrier to resistance and at 48 weeks in patients on entecavir or tenofovir which are highly potent with a higher genetic barrier to resistance.
• Virological breakthrough is a confirmed increase in HBV DNA level >1 log10 IU/ml compared with the nadir HBV DNA level on therapy. This usually precedes a biochemical breakthrough. The main causes of virological breakthrough are poor adherence to therapy or the development of resistance. Resistance may result in primary treatment failure or virological breakthrough on therapy.
• A sustained off-treatment virological response is defined as HBV DNA levels <2 000 IU/ml for at least 12 months after completion of treatment.
Histological response is a >2 points (histological activity index (HAI)) decrease in necroinflammatory activity without further progression in fibrosis compared to the pre-treatment histology.
Complete response is a sustained off-treatment virological response together with loss of HBsAg.
7. Available therapies
Six drugs are currently available in SA for treating chronic hepatitis B:
• Standard interferon alpha-2a and -2b
• Pegylated interferon alpha-2a
• Lamivudine
• Entecavir
• Tenofovir, which is available for off-label use alone or in combination with emtricitabine (it is not yet registered for the treatment of hepatitis B in SA, but is registered by both the European regulatory authority and the FDA for hepatitis B).
The recommended first-line monotherapies include interferon-based therapy and the NUCs tenofovir and entecavir. It is important to realise that all patients with chronic hepatitis B may be potential candidates for treatment, but it is important to choose the appropriate treatment at the appropriate time.
8. Indications for treatment
The 2008 NIH guideline'81 regarding indications for hepatitis B treatment suggests the following:
8.1 Patients who must be treated[6,20,29-33]
• Acute liver failure - in order to suppress ongoing HBV replication in an attempt to prevent ongoing hepatocyte necrosis
• Decompensated cirrhosis
• Advanced fibrosis or cirrhosis and detectable serum HBV DNA, even if normal ALT
• Patients receiving chemotherapy, rituximab or immuno-suppressive therapy.
8.2 Patients who should be considered for therapy[5,6,7]
• HBeAg-positive chronic hepatitis B
• HBeAg-negative chronic hepatitis B.
The decision to treat these patients and the type of treatment is clinically based on the combination of:
• Serum aminotransferase levels
• Serum HBV DNA levels
• HBV genotype and HBV mutations
• Histological grade and stage of the disease
• Race and age of the patient
• Family history of hepatitis B-related cirrhosis and HCC
• Co-factors such as alcohol, iron overload, and co-infection with either the hepatitis C virus (HCV) or HIV.
8.3 Patients who do not require immediate therapy[6]
• Patients in the immune tolerant phase (<30 years with persistently normal ALT, no evidence of liver disease, and no family history of cirrhosis or HCC)
• Patients in the inactive carrier or latency phase (if HBV DNA >2 000 and <20 000 IU/ml and no evidence of liver disease, monitor ALT every 3 months and HBV DNA every 6 - 12 months for 3 years, then lifelong as for inactive carriers)
• Occult hepatitis B.
9. Treatment of chronic hepatitis B
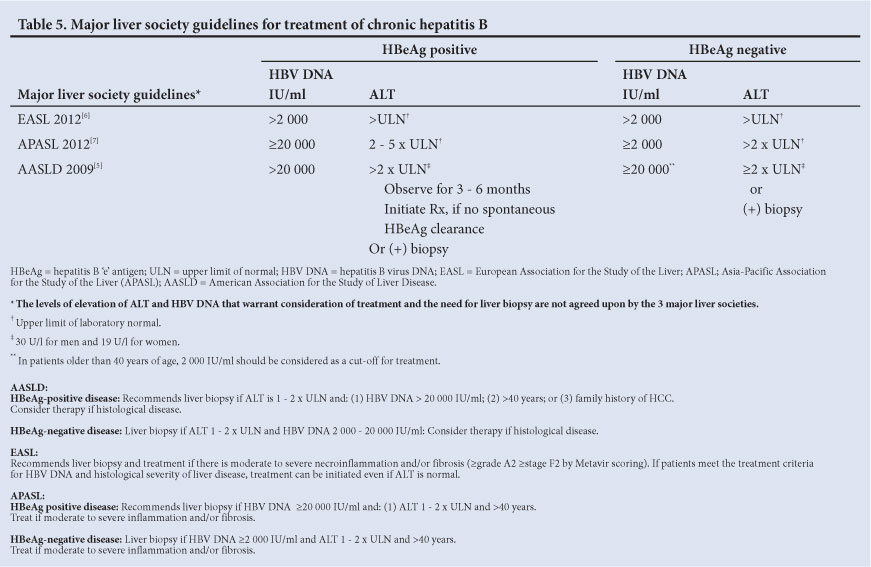
Over the past 4 years, the 4 major liver societies - the AASLD, EASL, APASL and WGO - have published their updated guidelines on the management of HBV infection.[5-7,9]
9.1 Treatment criteria for chronic hepatitis B
The AASLD, EASL and APASL all recommend slightly different HBV
DNA and ALT levels for initiation of treatment (Table 5). However,
it is important to treat liver disease and not only base the need for therapy on ALT and HBV DNA levels. In SA, patients frequently have a long immune tolerant phase and ALT is frequently <2 x the upper limit of normal (ULN) with high HBV DNA levels. Therefore, liver biopsy plays an important role in determining the need for therapy and the type of therapy. If the liver biopsy shows moderate to severe necro-inflammation and/or fibrosis using a standardised scoring system (>A2 or >F2 on Metavir scoring), patients should be considered for treatment. Other important factors determining the need for therapy and type of therapy include patient's age and race, potential future pregnancies in females, family history of HCC, co-factors (alcohol, iron overload and viral co-infection) and anticipated compliance with potentially lifelong NUC therapy. Antiviral therapy may also need to be considered in healthcare workers without active liver disease if they are engaged in exposure-prone procedures, in order to suppress viral replication. It is important to continue monitoring all patients with chronic HBV infection who are not on therapy as they may require treatment in the future.
See SA treatment algorithms for chronic hepatitis B (HBeAg-positive and HBeAg-negative disease) (Figs 1 and 2).

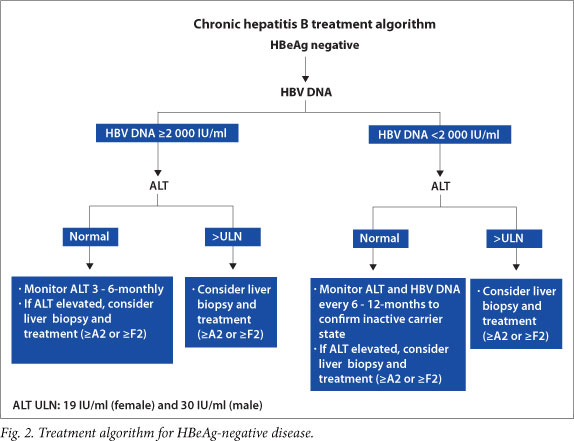
10. Management strategies
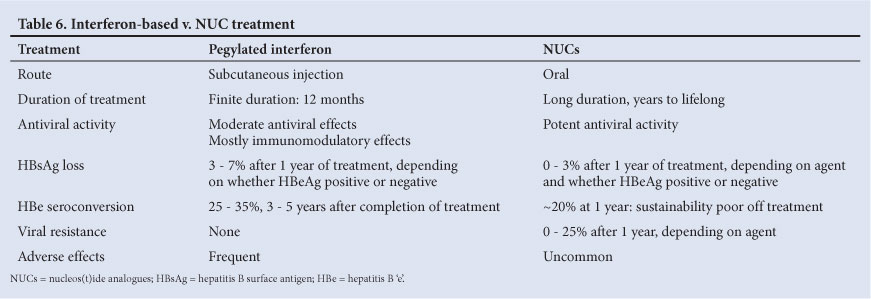
Once a decision has been made to treat a patient with chronic hepatitis B, all the pros and cons of interferon-based therapy v. NUCs need to be considered and discussed with the patient (Table 6).
10.1 General measures
All patients should be screened for IgG anti-HAV, anti-HCV and anti-HIV before treatment is initiated. Those found not to be immune to hepatitis A should be vaccinated.
10.2 Adverse effects of interferon-based therapy
• Initial influenza-like illness
• Fatigue
• Anorexia and weight loss
• Alopecia
• Myelosuppression with neutropenia and thrombocytopenia
• Hypo- and hyper-thyroidism.
• Emotional lability and depression
• Retinal changes and impaired vision
• Flare in ALT occurs in 30 - 40% of patients on treatment.
The main advantages of interferon alpha-based treatment are the lack of resistance, the finite duration of therapy, the possibility of immune-mediated clearance of HBV and the slightly higher probabilities of HBeAg to anti-HBe and HBsAg to anti-HBs seroconversions rates after the 1st year of treatment. There is also a cumulative benefit, as ongoing HBeAg loss continues after cessation of therapy (25 - 35%, 3 - 5 years after therapy)[34] as does HBsAg loss.
10.3 Interferon alpha and pegylated interferon alpha therapy contraindications[5,16]
• Decompensated cirrhosis
• Fulminant hepatitis B
• Pregnancy
• Significant cardiopulmonary disease
• Uncontrolled seizures
• Active autoimmune disease
• Psychiatric disease
• Chemotherapy.
10.4 Factors favouring interferon alpha or pegylated interferon alpha as initial therapy[5,6]
10.4.1 Favourable predictors of response
10.4.1.1 Pretreatment
HBeAg-positive chronic hepatitis B (predictors of anti-HBe sero-conversion)
• Low viral load: HBV DNA <1 x 107 IU/ml. Note that definitions of low viral load vary from <2 x 106 to <2 x 108 IU/ml
• ALT >2 - 5 x ULN
• Active necroinflammation on biopsy (Metavir Grade >A2)
• Genotype A and B > C and D,[35-37] although choice of treatment should not be based on genotype alone.
HBeAg-negative chronic hepatitis B
• No strong pre-treatment predictors of virological response.
10.4.1.2 During treatment
HBeAg-positive chronic hepatitis B
• HBV DNA <20 000 IU/ml at 12 weeks of pegylated interferon alpha therapy is associated with a 50% chance of anti-HBe seroconversion.
• ALT flare followed by an HBV DNA decrease is associated with more frequent anti-HBe seroconversion.
• HBeAg decrease to <100 IU/ml at week 24 may predict anti-HBe seroconversion, if HBeAg quantification is available.[38]
• HBsAg level <1 500 IU/ml at week 12 ofpegylated interferon alpha therapy is associated with a 57% chance of sustained immune control (anti-HBe seroconversion 6 months post therapy).[39] HBsAg >20 000 IU/ml at week 12 is associated with low rate of anti-HBe seroconversion and treatment can be stopped.
HBeAg-negative chronic hepatitis B
• HBV DNA <20 000 IU/ml at 12 weeks of pegylated interferon alpha therapy is associated with a 50% chance of a sustained off-treatment response (normal ALT and HBV DNA <2 000
IU/ml)[38,40]
• Decreases in quantified HBsAg levels of >0.5 log10 IU/ml and >1 log10 IU/ml at weeks 12 and 24 of therapy, respectively, have high predictive values for undetectable HBV DNA 24 weeks post completion of treatment.[41]
10.4.2 Patient demographics[5,34]
Younger patients, and particularly young women wanting future pregnancies.
10.4.3 No co-infection with HIV
10.4.4 HCV co -infection with HBV
10.5 Factors favouring NUC as initial therapy
• High HBV DNA levels (>2 x 108 IU/ml)
• Patient demographics: older patients
• Ability to commit to potentially lifelong therapy
• HIV co-infection
• Contraindications to interferon-based therapy
• HBV genotype does not influence response to NUCs.
For both interferon-based and NUC therapy: low HBV DNA levels < 1 x 107 IU/ml, ALT >2 - 5 x ULN and active necroinflammation on biopsy are predictive of anti-HBe seroconversion.
10.6 Duration of treatment and dosage regimens[5,6]
10.6.1 Pegylated interferon alpha-2a
A 48-week course of 180 µg pegylated interferon alpha-2a, given subcutaneously weekly, should be considered as first-line therapy in both HBeAg-positive and HBeAg-negative disease in patients with a favourable pre-treatment profile (ALT >3 x ULN, HBV DNA
<2 x 106 IU/ml, genotype A and B and active necroinflammation on liver biopsy).
If the HBV DNA levels are detectable, but <2 000 IU/ml at 48 weeks, there is no need for ongoing NUC treatment unless the patient is cirrhotic or has >F3 fibrosis. However, ongoing monitoring is required. If the HBV DNA levels are >2 000 IU/ml at 48 weeks, ongoing treatment with NUCs is required.
10.6.2 Interferon alpha-2a or -2b
Interferon alpha-2a or -2b given subcutaneously 3 times a week for 16 - 24 weeks is less expensive than pegylated interferon alpha and is effective in carefully selected patients.
Potential suitable candidates are HBeAg positive with high baseline ALT levels, low HBV DNA levels and active necroinflammation on biopsy.
The dosage of interferon alpha can be titrated from 1 to 10 MU subcutaneously 3 times a week or 5 MU daily. In the SA setting, dosages seldom exceed 5 million units 3 times a week and are usually 3 million units 3 times a week. The recommended dosage for children is 6 MU/m2 3 times a week with a maximum dosage of 10 MU 3 times a week.
A liver biopsy is mandatory when considering treatment with either pegylated interferon alpha-2a or standard interferon alpha.
10.6.3 NUCs
A finite duration of treatment may be achievable in HBeAg-positive patients who achieve anti-HBe seroconversion and undetectable HBV DNA levels on treatment.
This is only possible with potent NUCs such as entecavir and tenofovir which have a high genetic barrier to resistance. Once HBeAg to anti-HBe seroconversion has occurred, treatment should be consolidated and continued for at least 1 year. Careful follow-up after the cessation of successful treatment is important as up to 20% of patients may relapse and become HBeAg positive again. Continuation of therapy until HBsAg seroconversion is advisable.[42]
Patients with HBeAg-negative chronic hepatitis B and those with cirrhosis require lifelong treatment with NUCs.
Lamivudine: The recommended dosage for adults with normal renal function (creatinine clearance >50 ml/min) is 100 mg/day. Dosage reduction is necessary in patients with impaired renal function. A practical issue is that given the availability and use of lamivudine in the treatment of HIV in SA, the use of the 150 mg tablet daily in the treatment of hepatitis B mono-infection is acceptable.
The recommended dosage for children is 3 mg/kg/day with a maximum dosage of 100 mg/day. A liquid formulation for children is available. Patients who are HIV/HBV co-infected should receive lamivudine 150 mg bd.
Tenofovir: The recommended dosage for adults with normal renal function (creatinine clearance >50 ml/min) is 300 mg per day. It is necessary to reduce the dosage in patients with impaired renal function.
Entecavir: The recommended dosage for adults with normal renal function (creatinine clearance >50 ml/min) is 0.5 mg daily if lamivudine naive and 1 mg daily if previously exposed to lamivudine or if lamivudine refractory or resistant. Dosage reduction is necessary in patients with impaired renal function.
11. Treatment of patients with compensated cirrhosis
Treatment should be considered in patients with compensated cirrhosis and detectable HBV DNA levels.[6,20,43]
Interferon-based therapy can be used in compensated cirrhosis, but does increase the risk of infections and decompensation. If interferon-based therapy is used, it is important to titrate the dose of standard interferon/pegylated interferon with careful monitoring of the FBC, differential count and ALT. The duration of therapy (16 - 24 weeks for standard interferon and 48 weeks for pegylated interferon) should be timed from the point that the maximum dosage was achieved during the titration.
NUCs are well tolerated and a potent NUC with a high genetic barrier to resistance should be used, e.g. tenofovir or entecavir. If lamivudine is used, it is advisable that it be combined with tenofovir. Long-term therapy is required and regular monitoring of HBV DNA levels is essential. Any decompensation on NUC therapy could be due to the natural progression of the disease or the development of HCC, but it is critical to first exclude non-compliance or the development of resistance as a factor. Current evidence suggests that prolonged and effective suppression of HBV DNA replication can stabilise and even prevent or delay the need for liver transplantation.[20] NUC therapy is usually lifelong.
12. Treatment of decompensated cirrhosis
All patients with decompensated cirrhosis should be considered for urgent treatment. Interferon-based therapy is contraindicated and only NUCs should be used.[5,6,8,16] Treatment is indicated even if the HBV DNA level is low or undetectable, in order to prevent flares/reactivation. Treatment with NUCs may lead to clinical improvement over a period of 3 - 6 months. If there is ongoing deterioration, treatment with NUCs is important to suppress the HBV DNA and thereby decrease the risk of hepatitis B recurrence post-liver transplantation. In unstable patients with deteriorating renal function, it is advisable to start with lamivudine and add in tenofovir once the clinical condition has stabilised. If entecavir is used, the recommended dosage is 1 mg daily and patients should be monitored for lactic acidosis. Lifelong treatment is recommended.
13. Treatment of patients in the inactive carrier state or the immune tolerant phase who require immunosuppressive therapy, rituximab or chemotherapy
• HBsAg and IgG anti-HBc should be tested before the introduction of immunosuppressive therapy, rituximab or chemotherapy.[44-46]
• If either HBsAg or IgG anti-HBc is positive, HBV DNA levels should be measured.
• If HBsAg negative, IgG anti-HBc positive and HBV DNA is detectable, NUC therapy is indicated as for HBsAg-positive patients.
• If HBsAg negative, IgG anti-HBc positive and HBV DNA is undetectable, no treatment is needed, but ALT and HBV DNA levels should be monitored at regular intervals (1 - 3-monthly) depending on immunotherapy type; treatment should be initiated when HBV DNA becomes detectable. If regular HBV DNA level monitoring is not possible, NUC therapy is also indicated.
• If HBsAg positive and HBV DNA <2 000 IU/ml, then treatment with an NUC should be continued for 12 months after completion of immunosuppressive therapy. Lamivudine can be used, if anticipated duration of treatment is not >12 months and
HBV DNA level is <2 000 IU/ml.
• If HBsAg positive and HBV DNA >2 000 IU/ml, an NUC with a high genetic barrier to resistance (tenofovir or entecavir) should be used and continued until the usual treatment endpoint has been achieved. Tenofovir or entecavir should also be used, if lengthy and repeated cycles of immunosuppression are needed.
• Where possible, antiviral therapy should be initiated before the onset of immunosuppressive therapy, rituximab or chemotherapy, and HBV DNA levels should be undetectable.[47]
• IgG anti-HBc-positive patients receiving bone marrow or stem cell transplants should also receive NUC prophylaxis.
14. Combination therapy
There are as yet no data confirming the advantage of combination therapy.
The most commonly used combination therapies are tenofovir plus lamivudine or tenofovir plus emtricitabine, which may be considered in the following situations:[6,48,49]
• Patients with high baseline HBV DNA levels who are at greater risk of developing resistance
• Cirrhotic patients in whom a biochemical breakthrough associated with the development of resistance is potentially life threatening
• Post-liver transplantation together with hepatitis B immune globulin (HBIG)
• HIV/HBV co-infection where there is a risk of resistance with monotherapy
• A suboptimal response to an initial drug, especially in the presence of high HBV DNA levels
• Established resistance to an NUC.
15. Monitoring therapy[5,6]
15.1 Pegylated interferon alpha (Table 7)
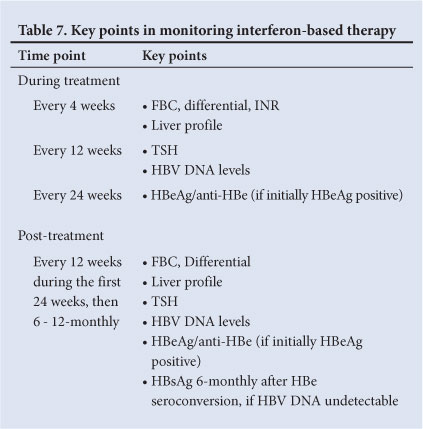
15.1.1. During treatment
• FBC, differential count, INR and liver profile should be performed at least monthly. The baseline results and effects of therapy may dictate more frequent testing.
• Serum HBV DNA levels should be measured at weeks 12 and 24 on treatment to assess the primary response and at end of treatment.
• Serum thyroid-stimulating hormone (TSH) every 12 weeks.
• Monitor for known side-effects of interferon.
15.1.2 Post treatment
• FBC, differential count, liver profile, TSH, HBV DNA levels,
HBeAg/anti-HBe (if initially HBeAg positive) should be measured every 12 weeks during the first 24 weeks post treatment and then 6 - 12-monthly
• FBC, differential count, liver profile and DNA levels may need more frequent monitoring if patient is unstable
• 6 - 12-monthly HBsAg monitoring.
HBeAg-positive disease: Aim for a sustained off-treatment anti-HBe seroconversion, ALT normalisation and HBV DNA <2 000
IU/ml.
Undetectable HBV DNA by real-time PCR is associated with increased chance of HBsAg loss:
• Test for HBeAg and anti-HBe at weeks 24 and 48 of treatment and 24 weeks post treatment. HBsAg should be checked 6 monthly after HBeAg to anti-HBe seroconversion if HBV DNA is undetectable.
• If a 1 log10 reduction in HBV DNA levels is not achieved at 12 weeks, the pegylated interferon alpha should be stopped and an NUC introduced.
HBeAg-negative disease: Aim for a sustained off-treatment virological response with HBV DNA levels <2 000 IU/ml and ALT normalisation.
Undetectable HBV DNA by real-time PCR is associated with increased chance of long-term HBsAg loss:
• Monitor HBsAg 6-monthly, if HBV DNA levels undetectable
• If a 1 log10 reduction in HBV DNA levels is not achieved at 12 weeks, the pegylated interferon alpha should be stopped and an NUC introduced.
15.2 NUC therapy (Table 8)
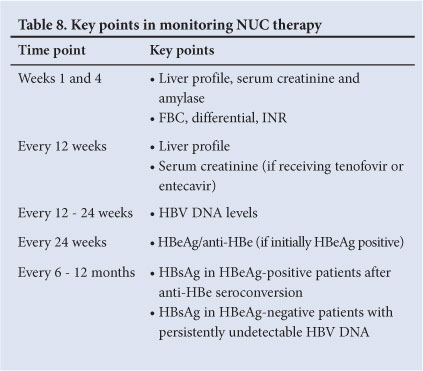
• FBC, differential count, INR, liver profile, amylase and creatinine assessment should be performed at weeks and 4 and then 3-monthly if stable.
• HBV DNA levels are measured at week 12 to assess virological response and then every 12 - 24 weeks.
• HBV DNA monitoring is critical to detect treatment failure.
Undetectable HBV DNA levels by real-time PCR (level of detection <10 - 15 IU/ml) need to be achieved to prevent the development of resistance.
• Partial responses (HBV DNA level detectable but <2 000 IU/ml) are assessed at 24 weeks for lamivudine and at 48 weeks for tenofovir and entecavir. If HBV DNA levels are still positive, but declining at 48 weeks on tenofovir or entecavir, monotherapy can be continued.[49]
• NUCs require dosage adjustments in the setting of renal impairment.
15.2.1 HBeAg-positive disease
• HBeAg and anti-HBe should be measured every 6 - 12 months. Consider stopping NUCs 48 weeks after HBeAg seroconversion; however, this must be carefully considered and if it is stopped, the patient must be monitored closely. In patients with cirrhosis or previous hepatic decompensation, NUCs should never be stopped.
• HBsAg should be checked 6-monthly after anti-HBe sero-conversion.
15.2.2 HBeAg-negative disease
• A virological response (HBV DNA < 2000 IU/ml) is associated with disease remission
• Monitor HBsAg 6-monthly, if HBV DNA levels are undetectable. (See Table 8 for key points for monitoring NUC therapy.)
16. Management of nucleos(t)ide resistance[5,6]
• Lamivudine resistance: Add tenofovir or switch to tenofovir/ emtricitabine. Screen for tyrosine-methionine-aspartate-aspartate (YMDD) mutations, if available.
• Entecavir resistance: Add or switch to tenofovir or switch to tenofovir plus emtricitabine. The safety of an entecavir/tenofovir combination is not known.
• Tenofovir: resistance has not yet been described.
17. Monitoring of patients not considered for therapy[5]
17.1 Immune tolerant phase (HBeAg positive, HBV DNA >20 000 IU/ml, normal ALT)
• ALT and HBV DNA levels every 3 - 6 months, more often if ALT becomes elevated.
• HBeAg status every 6 - 12 months.
• If ALT levels are 1 - 2 x ULN, recheck ALT every 1 - 3 months. Consider liver biopsy if the patient is >40 years of age, and if ALT is borderline or mildly elevated on serial tests. Consider treatment if biopsy shows moderate/severe inflammation or significant fibrosis.
• If ALT is >2 x ULN for 3 - 6 months, HBeAg positive and HBV DNA is >20 000 IU/ml, consider liver biopsy and treatment.
• Consider screening for HCC in the relevant population.
17.2 Inactive HBsAg carrier state (HBsAg positive, HBV DNA <2 000 IU/ml, normal ALT)
• Monitor ALT and HBV DNA levels every 3 - 4 months for 1 year.
• If ALT is persistently normal and HBV DNA <2 000 IU/ml, then check ALT and HBV DNA every 6 - 12 months.
• If ALT >1 - 2 x ULN, check serum HBV DNA level and exclude other causes of liver disease. Consider liver biopsy if ALT is borderline or mildly elevated on serial tests, or if HBV DNA is persistently >2 000 IU/ml. Consider treatment if biopsy shows moderate/severe inflammation or significant fibrosis.
• Consider screening for HCC in relevant populations.
18. Special patient populations[5,6]
18.1 Pregnancy
Interferon-based therapy is contraindicated in pregnancy and family planning should always be discussed before embarking on therapy.
Lamivudine and entecavir are category C drugs and tenofovir is a category B drug. Data in HIV-positive pregnant women suggest that the use of lamivudine, emtricitabine and tenofovir is safe.!50,511 There is evidence that in HBsAg-positive women with high levels of viraemia (HBV DNA >2 x 107 IU/ml), treatment with lamivudine during the last trimester reduces the risk of intra-uterine and perinatal transmission of HBV, when given in addition to HBIG and HBV vaccination at delivery.[52] It is now recommended that lamivudine or tenofovir should be used in the last trimester in HBsAg-positive women with high viral loads (serum HBV DNA >1 x 106-7 IU/ml), to prevent intra-uterine and perinatal HBV transmission. NUC therapy can be discontinued 3 months post-delivery if only required for prevention of perinatal transmission.
HBV-infected women should be monitored closely after delivery as flares may occur.[53]
18.2 Healthcare workers
Healthcare workers who are HBsAg positive and have HBV DNA levels >2 000 IU/ml should be treated with a potent antiviral agent with a high genetic barrier to resistance, such as tenofovir or entecavir. The HBV DNA level should preferably be undetectable or at least <2 000 IU/ml before such an individual may return to exposure-prone procedures.[6]
18.3 Children
Chronic hepatitis B is typically benign in children as they are usually in the immune tolerant phase. Only standard interferon alpha, lamivudine and adefovir have been evaluated in children.!54,551
In children with abnormal liver profiles, one should be guided by the histology in determining the need for treatment. In SA the choice is between standard interferon alpha or lamivudine therapy. Long-term use of lamivudine is associated with the development of resistance (70% at 5 years).
18.4 Dialysis and renal transplant patients
The dosages of lamivudine, tenofovir and entecavir need to be carefully adjusted in patients with impaired renal function. Tenofovir or entecavir can be used in renal transplant patients and the dosage adjusted according to the renal graft function. Interferon-based therapy is not recommended in renal transplant recipients because of the risk of graft rejection. All HBsAg-positive patients undergoing renal transplantation should receive prophylactic NUC therapy.
18.5 Extrahepatic disease
Patients with chronic hepatitis B and active HBV replication who present with extrahepatic disease (polyarteritis nodosa, glomerulo-nephritis) should be considered for therapy with NUCs, but efficacy is variable. Lamivudine has been most widely used, but tenofovir or entecavir are now preferable, provided renal function permits their use. Plasmapheresis and steroids, in combination with an NUC, have been used in the initial phase of treatment. Interferon-based therapy may worsen immune-mediated extra-hepatic manifestations.
18.6 HBV/HCV co-infected patients
HCV co-infection accelerates the progression of liver disease and increases the risk of HCC. However, in co-infected patients, HBV DNA levels tend to be low and the hepatitis C virus is usually responsible for disease activity. Liver biopsy and HBV DNA levels are useful in establishing the contribution of hepatitis B to disease activity. The patient should be treated with pegylated interferon alpha and ribavirin, as indicated for chronic hepatitis C.!561 Sustained virological responses are similar to reponses in those who are mono-infected.!57-591 In individuals with chronic hepatitis C genotype 2 and 3, if the chronic hepatitis B requires treatment, then pegylated interferon alpha could be continued for 1 year or NUC therapy considered. There is a potential risk of HBV reactivation during treatment or after HCV clearance, which should be treated with NUCs.
18.7 HBV/HIV co-infected patients
HBV/HIV co-infected patients are at greater risk of progression to cirrhosis and have a higher risk ofHCC.[3,60-62] As immune reconstitution, following the initiation of antiretrovirals, can lead to potentially life-threatening flares of hepatitis B, all HBV/HIV co-infected patients with a CD4 count <350 cells/ml should receive antiretroviral therapy that is also active against the HBV. Therapy should include lamivudine, tenofovir or tenofovir/emtricitabine, together with a third agent active against HIV.!631 If antiretrovirals need to be changed because of HIV resistance or drug toxicity, then tenofovir and lamivudine or tenofovir/ emtricitabine should be continued together with the new antiretroviral drugs. Pegylated interferon is only considered in patients who have a CD4 count >500 cells/ml.!161
18.8 Severe acute hepatitis B
Antiviral therapy is not necessary for uncomplicated symptomatic acute hepatitis B, as >95% of immunocompetent adults will spontaneously clear HBV. It has been reported that lamivudine improves survival in patients with severe or fulminant hepatitis B.[30,32] Treatment of these patients is also justified, as reducing HBV DNA to undetectable levels lowers the risk of recurrent hepatitis B, should they require liver transplantation. NUC therapy has been recommended in patients with prolonged, severe acute hepatitis B (elevated INR >1.5 and marked jaundice persisting for longer than 4 weeks).[11] It is also recommended that elderly or immunosuppressed patients and those co-infected with HCV should be treated, as they are more likely to have a subfulminant/fulminant course.
Lamivudine can be used in the acute setting as treatment is usually of short duration, unless liver transplantation is required. Entecavir can also be used. However, tenofovir has the potential for nephrotoxicity and should be used with caution when the patient's clinical condition is unstable in the acute setting. NUC therapy should be continued for at least 3 months after seroconversion to anti-HBs, or 12 months after anti-HBe seroconversion without HBsAg loss, or indefinitely if the patient undergoes liver transplantation.
Interferon-based therapy is contraindicated because of the risk of acute liver failure.
19. Prevention of hepatitis B
19.1 Prevention of transmission of hepatitis B from individuals with chronic HBV infection
Patients with chronic hepatitis B should receive counselling regarding cofactors likely to accelerate disease progression (such as alcohol), the risk and modes of transmission and the need for long-term follow-up.
The following is advised:
• Abstinence. Significant ethanol intake (>20 g/day in women and >30 g/day in men) is associated with an increased risk of development of cirrhosis.[64,65]
• Household members and sexual partners are at increased risk of HBV infection and should be vaccinated if they are HBsAg, anti-HBs and IgG anti-HBc negative.
• Individuals who are HBsAg positive should:
• Use barrier protection during sexual intercourse if the partner is neither immune nor has been vaccinated
• Not share razors or toothbrushes
• Not donate blood, organs or sperm
• Follow standard universal precautions with open cuts or bleeding
• Inform their dentist of their HBV status.
19.2 Post-exposure prophylaxis
19.2.1 Needlestick injury/sexual exposure/mucosal or percutaneous (bite) exposure
• Wounds should be washed with soap and water, and mucous membranes flushed with water.
• Source individual should be screened for HBsAg, HIV and HCV Ab.
• Check HBsAg, anti-HBs and IgG anti-HBc in the exposed individual, to assess whether the individual is infected, immune or non-immune to hepatitis B.
• If source individual is HBsAg positive or status is unknown, give HBIG (0.06 ml/kg or 500 IU) intramuscularly and commence active vaccination (0, 1 and 2 months) if exposed individual is non-immune. HBIG and vaccine to be given at different injection sites. Repeat HBIG at 1 month, if the contact is HBeAg positive, has high HBV DNA levels or if this information is not known. If the exposed individual is a known non-responder to HBV vaccination, then 2 doses of HBIG should be given 1 month apart.
• Anti-HBs titres should be measured 1 - 2 months after vaccination.
19.2.2 Babies born to HBsAg-positive mothers
• HBIG (200 IU IM) and hepatitis B vaccine should be administered at different sites within 12 hours of delivery.[66] The vaccine and immunoglobulin must be given at different injection sites. Thereafter, the same immunisation schedule is followed as for other infants, with additional doses of HBV vaccine given at 6, 10 and 14 weeks according to the South African EPI.
• Ideally the active and passive immunisation should be given within 24 hours of delivery (preferably <12 hours), but immunisation is probably protective if administered up to 72 hours after delivery.
• The combination of active and passive immunisation is 95% effective in preventing perinatal transmission, but this is probably lower if the maternal HBV DNA levels are >2 x 107 IU/ml.
• If the mother is HBeAg positive or has high HBV DNA levels, HBIG can be repeated at 1 month.
• HBsAg and anti-HBs titres should be measured at 9 - 18 months of age. If anti-HBs titres are <10 mIU/ml, a second course of vaccination should be given. If HBsAg positive, the infant should be referred for further monitoring.
19.2.3 Prevention of recurrent hepatitis B after liver transplantation
Pre-transplant therapy with a potent NUC and a high genetic barrier to resistance is recommended for all HBsAg-positive patients undergoing liver transplantation, in an attempt to achieve an undetectable HBV DNA level before transplantation.[67-70] Post transplantation, an NUC must be used in combination with HBIG. To date, lamivudine and/or adefovir have been used in combination with HBIG and this has reduced the risk of recurrent hepatitis B to <10%. However, the more potent NUCs with lower risks of resistance (entecavir, tenofovir) or combination NUC therapy (lamivudine and tenofir or tenofovir/emtricitabine) should now be used together with HBIG. Lifelong antiviral therapy to prevent recurrent hepatitis B is required. The dosage, mode of administration (IVI or IMI) and duration of HBIG therapy in combination with potent NUCs are not yet established.
19.2.4 Prevention of hepatitis B following transplantation of non-hepatic organs from donors who are HBsAg negative and IgG HBcAb positive
• Risk of infection is low, ranging from 0 to 13%[71]
• Should ideally be given to an HBV-immune recipient
• If the recipient is HBV seronegative, antiviral therapy should be given to prevent de novo hepatitis B, particularly if the donor is
HBV DNA positive
• Optimal duration of prophylactic treatment is not known, but 6 - 12 months might be sufficient.
19.2.5 Prevention of hepatitis B following the transplantation of livers from donors who are HBsAg negative and IgG HBcAb positive
• Risk of de novo hepatitis B infection is as high as 75%[72-74 depending on the HBV immune status of the recipient. The risk is particularly high in endemic countries such as SA, where these donors often have occult hepatitis B.
• Lifelong antiviral therapy is recommended.[75]
• In the setting of potent NUCs with a high barrier to resistance, the need for, optimal dosage and duration of HBIG prophylaxis is currently not known.
• At present, we recommend a combination of HBIG and antiviral therapy (tenofovir +/- lamivudine).
19.3 Vaccination
Since April 1995, HBV vaccination has been part of the South African EPI and is given at 6, 10 and 14 weeks of age. If a dosage is missed, then catch-up doses are given 1 month apart to complete the schedule. Vaccination is recommended in the following individuals:
i. All infants, through the EPI ii. Infants and adolescents not previously vaccinated should receive catch-up vaccination iii. I ndividuals at increased risk of HBV infection as a result of percutaneous or mucosal exposure to blood or blood products, as well as those at risk of more severe infection. These include:
• Healthcare personnel including student healthcare workers and domestic workers in healthcare facilities
• Laboratory staff working with clinical specimens
• Policemen, firemen and members of the armed forces
• Personnel and residents of the correctional services and institutions/schools for the mentally handicapped
• All personnel and children attending creches and preschools
• Morticians and embalmers
• IV drug users
• Men who have sex with men
• Patients in haemodialysis or oncology units
• Transplant candidates before transplantation
• Household contacts and sexual partners of HBsAg-positive individuals
• Individuals receiving frequent blood or blood product transfusions
• Individuals with HIV or chronic hepatitis C.
It is important to remember that hepatitis B is endemic in SA. Thus all South Africans are potentially at risk of contracting hepatitis B infection and should consider vaccination.
HBV vaccines are either recombinant or plasma derived. Both formulations are safe and do not transmit hepatitis B or HIV. Plasma-derived vaccines are thought to be more immunogenic. Dosing schedules depend on the type of vaccine, age of administration, need for rapid immunisation and previous non-response to HBV vaccination. Combined hepatitis A and B vaccines are also available. Approximately 10% of healthy adults do not mount an anti-HBs response (>10 mIU/ml) to the primary immunisation schedule and should receive a repeat 3-dose (1 month apart) vaccination. This gives rise to protective antibody levels in 44 - 100% of individuals. Individuals who do not develop protective HBs antibody levels 1 - 2 months after revaccination can be considered for repeat vaccination (0, 1 and 2 months with a 6-month booster) with double the standard dosage of vaccine.
20. Screening for HCC[5,9,76]
The aim of HCC screening is to detect tumours smaller than 3 cm and preferably less than 2 cm in order to offer curative therapy. Cirrhotics have the highest risk of HCC. However, in SA, HBV infection is frequently acquired in childhood, with the consequent risk of occult infection and HBV gene incorporation into the hepatocyte genome, and HCC can therefore develop in a non-cirrhotic liver.
HCC surveillance with ultrasound of the liver and serum α-fetoprotein is recommended every 6 - 12 months for:
• Africans older than 20 years
• Asian males >40 years and Asian females >50 years
• All cirrhotic patients regardless of age
• Individuals with a family history of HCC, regardless of age
• Any individual with HBV/HIV co-infection
• Individuals with HBeAg-positive or HBeAg-negative disease
• Any carrier aged >40 years with persistent or intermittent ALT elevation and/or a high HBV DNA level >2 000 IU/ml
• Any carrier who has other risk factors for HCC.
References
1. Vardas E, Mathai M, Blaauw D, et al. Preimmunization epidemiology of Hepatitis B virus infection in South African children. J Med Virology 1999;58:111-115. http://dx.doi.org/10.1002/(SICI)1096-9071(199906)58:2<111::AID-JMV2>3.0.CO;2-B1 [ Links ]
2. Huang K, Lin S. Nationwide vaccination: A success story in Taiwan. Vaccine 2000;18:S35-38. http:// dx.doi.org/10.1016/S0264-410X(99)00460-01 [ Links ]
3. Puoti M, Torti C, Bruno R, Filice G, Carosi G. Natural history of chronic Hepatitis B in co-infected patients. J Hepatology 2006; 44: S65-S70. http://dx.doi.org/10.1016/j.jhep.2005.11.015; [ Links ]
4. McMahon BJ. The natural history of chronic hepatitis B virus infection. Hepatology 2009; 49:S45-S55. http://dx.doi.org/10.1002/hep.228981 [ Links ]
5. Lok ASF, McMahon BJ. AASLD Practice Guidelines. Chronic Hepatitis B: Update 2009. Hepatology 2009;50(3):1-36. [ Links ]
6. European Association for the Study of the Liver. EASL Clinical Practice Guidelines: Management of chronic hepatitis B virus infection. J Hepatol 2012;57:167-185. [ Links ]
7. Liaw YF, Kao JH, Piratvisuth T et al. Asian-Pacific consensus statement on management of chronic Hepatitis B: A 2012 update - Asian-Pacific Association for the Study of the Liver (APASL). Hepatol Int 2012;6:531-561. [ Links ]
8. Sorrell MF, Belongia EA, Costa J, et al. National Institutes of Health Consensus Development Conference Statement: Management of hepatitis B. Ann Intern Med 2009;150:104-110. http://dx.doi. org/10.1002/hep.229461 [ Links ]
9. World Gastroenterology Organisation Practice Guideline: Hepatitis B. September 2008. http:// worldgastroenterology.org/hepatitis-b.html (accessed 11 April 2013). [ Links ]
10. Lavanchy D. Hepatitis B virus epidemiology, disease burden, treatment, and current and emerging prevention and control measures. J Viral Hepat 2004;11(2):97-107. !http://dx.doi.org/10.1046/j.1365-2893.2003.00487.x1 [ Links ]
11. Hoofnagle JH, Doo E, Liang TJ, Fleischer R, Lok AS. Management of Hepatitis B: Summary of a clinical research workshop. Hepatology 2007;45(4):1056-1075. !http://dx.doi.org/10.1002/hep.216271 [ Links ]
12. McMahon BJ, Alward WL, Hall DB, et al. Acute hepatitis B virus infection: Relation of age to the clinical expression of disease and subsequent development of the carrier state. J Infect Dis 1985;151(4):599-603. http://dx.doi.org/10.1093/infdis/151.4.5991 [ Links ]
13. Wieland SF, Chisari FV. Stealth and cunning: Hepatitis B and C viruses. J Virol 2005;79(15):9369-9380. !http://dx.doi.org/10.1128/JVI.79.15.9369-9380.20051 [ Links ]
14. Yim HJ, Lok AS. Natural history of chronic hepatitis B infection: What we knew in 1981 and what we know in 2005. Hepatology 2006;43(2):S173-S181. !http://dx.doi.org/10.1002/hep.209561 [ Links ]
15. Jacobson IM, Martin P, Schiff ER, Tobias H. A treatment algorithm for management of chronic Hepatitis virus infection in the United States: 2008 update. Clin Gastroenterol Hepatol 2008;6(12):1315-1341. !http://dx.doi.org/10.1016/j.cgh.2008.08.0211 [ Links ]
16. Lok AS, McMahon BJ. Chronic Hepatitis B. Hepatology 2007;45(2):507-539. http://dx.doi.org/10.1002/hep.215131 [ Links ]
17. Hadziyannis SJ, Papatheodoridis GV. Hepatitis B e antigen-negative chronic Hepatitis B: Natural history and treatment. Semin Liver Dis 2006;26(2):130-141. http://dx.doi.org/10.1055/s-2006-9397511 [ Links ]
18. Keefe EB, Dieterich DT, Han SH, et al. A treatment algorithm for management of chronic Hepatitis virus infection in the United States. Clin Gastroenterol Hepatol 2004;2(2):87-106. http://dx.doi.org/10.1016/S1542-3565(03)00312-41 [ Links ]
19. Colloredo MG, Leandro G, Brunetto MR, et al. Role of IgM antibody to hepatitis B core antigen in the diagnosis of hepatitis B exacerbations. Arch Virol 1993;8:203-211. [ Links ]
20. Liaw YF, Sung JJ, Chow WC, et al. Lamivudine for patients with chronic Hepatitis B and advanced liver disease. N Engl J Med 2004;351:1521-1531. !http://dx.doi.org/10.1056/NEJMoa0333641 [ Links ]
21. Chen CJ, Iloeje UH, Yang H. Long-term outcomes in Hepatitis B: The REVEAL-HBV study. Clin Liver Dis 2007;11:797-816. !http://dx.doi.org/10.1016/j.cld.2007.08.0051 [ Links ]
22. Iloeje UH, Yang HI, Su J, Jen CL, You SL, Chen CJ. Predicting cirrhosis risk based on the level of circulating Hepatitis B viral load. Gastroenterology 2006;130(3):678-686. http://dx.doi.org/10.1053/j. gastro.2005.11.0161 [ Links ]
23. Chen CJ, Yang HI, Su J, et al. Risk of hepatocellular carcinoma across a biological gradient of serum hepatitis B virus DNA level. JAMA 2006;295(1):65-73. !http://dx.doi.org/10.1001/jama.295.1.651 [ Links ]
24. Keefe EB, Dieterich DT, Han SH, et al. A treatment algorithm for management of chronic Hepatitis B virus infection in the United States: An update. Clin Gastroenterol Hepatol 2006;4(8):936-962. http:// dx.doi.org/10.1016/j.cgh.2006.05.0161 [ Links ]
25. Seeger C, Mason WS. Hepatitis B virus biology. Microbiol Mol Biol Rev 2000;64(1):51-68. http:// dx.doi.org/10.1128/MMBR.64.1.51-68.20001 [ Links ]
26. Werle-Lapostolle B, Bowden S, Locarnini S, et al. Persistence of cccDNA during the natural history of chronic hepatitis B and decline during adefovir dipivoxil therapy. Gastroenterology 2004;126(7):1750-1758. http://dx.doi.org/10.1053/j.gastro.2004.03.0181 [ Links ]
27. Brunetto MR. A new role for an old marker, HBsAg. J Hepatol. 2010;52(4):475-477. http://dx.doi. org/10.1016/j.jhep.2009.12.0201 [ Links ]
28. Marcellin P, Gane E, Buti M, et al. Regression of cirrhosis during treatment with tenofovir disoproxil fumarate for chronic hepatitis B: A 5-year open-label follow-up study. Lancet 2013;381(9865):468-475. [ Links ]
29. Degertekin B, Lok AS. Indications for therapy in Hepatitis B. Hepatology 2009;49:S129-S137. http://dx.doi.org/10.1002/hep.229311 [ Links ]
30. Kondili LA, Osman H, Mutimer D. The use of lamivudine for patients with acute hepatitis B (a series of cases). J Viral Hepat 2004;11(5):427-431. http://dx.doi.org/10.1111/j.1365-2893.2004.00504.x; [ Links ]
31. Loomba R, Rowley A, Wesley R, et al. Systematic review: The effect of preventive lamivudine on hepatitis B reactivation during chemotherapy. Ann Intern Med 2008;148(7):519-528. [ Links ]
32. Tilmann HL, Hadem J, Leifeld L, et al. Safety and efficacy of lamivudine in patients with severe acute or fulminant hepatitis B, a multicentre experience. J Viral Hepat 2006; 13(4): 256-263. !http://dx.doi.org/10.1111/j.1365-2893.2005.00695.x1 [ Links ]
33. Hoofnagle JH. Reactivation of Hepatitis B. Hepatology 2009;49:S156-S165. http://dx.doi.org/10.1002/hep.229451 [ Links ]
34. Lok AS. Drug therapy: Tenofovir. Hepatology 2010;52(2):743-747. http://dx.doi.org/10.1002/ hep.237881 [ Links ]
35. Janssen HL, van Zonneveld M, Senturk H, et al Pegylated interferon alfa-2b alone or in combination with lamivudine for HBeAg-positive chronic hepatitis B: A randomised trial. Lancet 2005;365(9454):123-129.http://dx.doi.org/10.1016/S0140-6736(05)17701-01 [ Links ]
36. Lau GK, Pirattvisuth T, Luo KX, et al. Peginterferon alfa-2a, lamivudine and the combination for HB eAg-positive chronic Hepatitis B. N Engl J Med 2005;352(26):2682-2695. [http://dx.doi.org/10.1056/ NEJMoa043470] [ Links ]
37. Flink HJ, van Zonneveld M, Hansen BE, de Man RA, Schalm SW, Janssen HL. Treatment with peginterferon alpha-2b for HB eAg-positive chronic hepatitis B: HB sAg loss is associated with HBV genotype. Am J Gastroenterol 2006;101(2):297-303. [http://dx.doi.org/10.1111/j.1572-0241.2006.00418.x] [ Links ]
38. Fried MW, Pirattvisuth T, Lau GK, et al HB eAg and Hepatitis B virus DNA as outcome predictors during therapy with peginterferon alfa-2a for HB eAg-positive chronic Hepatitis B. Hepatology 2008;47(2):428-434. [http://dx.doi.org/10.1002/hep.22065] [ Links ]
39. Sonneveld MJ, Rijckborst V, Boucher C, Hansen BE, Janseen HL. Prediction of sustained response to peginterferon alfa-2b for HBeAg-positive chronic hepatitis B using on-treatment hepatitis B surface antigen decline. Hepatology 2010;52(4):1251-1257. [http://dx.doi.org/10.1002/hep.23844] [ Links ]
40. Bonino F, Marcellin P, Lau GK, et al Predicting response to peginterferon {alpha}-2a, lamivudine and the two combined for HB eAg-negative chronic Hepatitis B. Gut 2007;56(5):699-705. [http://dx.doi.org/10.1136/gut.2005.089722] [ Links ]
41. Moucari R, Mackiewicz V, Lada O, et al. Early serum HB sAg drop: A strong predictor of sustained virological response to pegylated interferon alfa-2a in HB eAg-negative patients. Hepatology 2009;49(4):1151-1157. [http://dx.doi.org/10.1002/hep.22744] [ Links ]
42. Reijnders JG, Perquin MJ, Zhang N, Hansen BE, Janssen HL. Nucleos(t)ide analogues only induce temporary hepatitis B e antigen seroconversion in most patients with chronic hepatitis B. Gastroenterology 2010;139(2):491-498. [http://dx.doi.org/10.1053/j.gastro.2010.03.059] [ Links ]
43. Peters MG. Special populations with Hepatitis B virus infection. Hepatology 2009;49:S146-S155.[http://dx.doi.org/10.1002/hep.22965] [ Links ]
44. Hanbali A, Khaled Y. Incidence of Hepatitis B reactivation following Rituximab therapy. Am J Hematol 2009;84(3):195. [http://dx.doi.org/10.1002/ajh.21343] [ Links ]
45. Lalazar G, Rund D, Shouval D. Screening, prevention and treatment of viral hepatitis B reactivation in patients with haematological malignancies. Br J Hematol 2007;136(5):699-712. [http://dx.doi.org/10.1111/j.1365-2141.2006.06465.x] [ Links ]
46. Mindikoglu AL, Regev A, Schiff ER. Hepatitis B virus reactivation after cytotoxic chemotherapy: The disease and its prevention. Clin Gastroenterol Hepatol 2006;4(9):1076-1081. [http://dx.doi.org/10.1016/j.cgh.2006.05.027] [ Links ]
47. Liang R. How I treat and monitor viral Hepatitis B infection in patients receiving intensive immunosuppressive therapies or undergoing hematopoietic stem cell transplantation. Blood 2009;113(14):3147-3153. [http://dx.doi.org/10.1182/blood-200810-163493] [ Links ]
48. Lok AS, Zoulim F, Locarnini S, et al. Antiviral drug-resistant HBV: Standardization of nomenclature and assays and recommendation for management. Hepatology 2007;46(1):254-265. [http://dx.doi.org/10.1002/hep.21698] [ Links ]
49. Keeffe EB, Dieterich DT, Han SH, et al. A treatment algorithm for the management of chronic Hepatitis B virus infection in the United States: 2008 update. Clin Gastroenterol Hepatol 2008;6(12):1315-1341. [http://dx.doi.org/10.1016/j.cgh.2008.08.021] [ Links ]
50. Terrault NA, Jacobson IM.Treating chronic hepatitis B in patients who are pregnant or are undergoing immunosuppressive chemotherapy. Semin Liver Dis 2007;27:18-24. [http://dx.doi. org/10.1055/s-2007-984696] [ Links ]
51. Chotiyaputta W, Lok AS. Role of antiviral therapy in the prevention of perinatal transmission of hepatitis B virus infection. J Viral Hepat 2009;16(2):91-93. [http://dx.doi.org/10.1111/j.1365-2893.2008.01067.x] [ Links ]
52. van Zonneveld M, van Nunen AB, Niesters HG, de Man RA, Schalm SW, Janssen HL. Lamivudine therapy during pregnancy to prevent perinatal transmission of hepatitis B virus infection. J Viral Hepat 2003;10(4):294-297. [http://dx.doi.org/10.1046/j.1365-2893.2003.00440.x] [ Links ]
53. ter Borg MJ. Leemans WF, de Man RA, Janssen HL. Exacerbation of chronic hepatitis B infection after delivery. J Viral Hepat 2008;15(1):37-41. [http://dx.doi.org/10.1111/j.1365-2893.2007.00894.x] [ Links ]
54. Jonas MM, Little NR, Gardner SD. Long-term lamivudine treatment of children with chronic hepatitis B: Durability of therapeutic responses and safety. J Viral Hepat 2008;15(1):20-27. [http://dx.doi. org/10.1111/j.1365-2893.2007.00891.x] [ Links ]
55. Jara P, Bortolottti F. Interferon-alpha treatment of chronic hepatitis B in childhood: A consensus advice based on experience in European children. J Pediatr Gastroenterol Nutr 1999;29(2):163-170. [http://dx.doi.org/10.1097/00005176-199908000-00012] [ Links ]
56. Potthoff A, Wedemeyer H, Boecher WO, et al. The HEP-NET B/C co-infection trial: A prospective multicenter study to investigate the efficacy of pegylated interferon alpha-2b and ribavirin in patients with HBV/HCV co-infection. J Hepat 2008;49(5):688- 694. [http://dx.doi.org/10.1016/j. jhep.2008.03.028] [ Links ]
57. Chu CJ, Lee SD. Hepatitis B virus/hepatitis C virus coinfection: Epidemiology, clinical features, viral interactions and treatment. J Gastroenterol Hepatol 2008;23(4):512-520. [http://dx.doi.org/10.1111/ j.1440-1746.2008.05384.x] [ Links ]
58. Liu CJ, Chen PJ, Lai MY, Kao JH, Jeng YM, Chen DS. Ribavirin and interferon is effective for hepatitis C virus clearance in hepatitis B and C dually infected patients. Hepatology 2003;37(3):568-576. [http://dx.doi.org/10.1053/jhep.2003.50096] [ Links ]
59. Zhou J, Dore GJ, Zhang F, Lim PL, Chen YMA. Hepatitis B and C virus coinfection in the TREAT Asia HIV observational database. J Gastroenterol Hepatol 2007;22(9):1510-1518. [http://dx.doi. org/10.1111/j.1440-1746.2007.05062.x] [ Links ]
60. Hoffman CJ, Thio CL. Clinical implications of HIV and hepatitis B co-infection in Asia and Africa. Lancet Infect Dis 2007;7(6):402-409. [http://dx.doi.org/10.1016/S1473-3099(07)70135-4] [ Links ]
61. Soriano V, Puoti M, Bonacini M, et al. Care of patients with chronic hepatitis B and HIV co-infection: Recommendations from an HIV-HBV International Panel. AIDS 2005;19(3):221-240. [http://dx.doi.org/10.1097/01.aids.0000163948.62176.e7] [ Links ]
62. Sulkowski MS. Viral hepatitis and HIV Co-infection. J Hepatol 2008;48(2):353-367. [http://dx.doi. org/10.1016/j.jhep.2007.11.009] [ Links ]
63. Benhamou Y, Fleury H, Trimoulet P, et al Anti-hepatitis B virus efficacy of tenofovir disoproxil fumarate in HIV-infected patients. Hepatology 2006;43(3):548-555. [http://dx.doi.org/10.1002/hep.21055] [ Links ]
64. Chevillotte G, Durbec JP, Gerolami A, Berthezene P, Bidart JM, Camatte R. Interaction between hepatitis B virus and alcohol consumption in liver cirrhosis: An epidemiologic study. Gastroenterology 1983;85(1):141-145. [ Links ]
65. Villa ERL, Barchi T, Ferretti I, et al. Susceptibity of chronic symptomless HBsAg carriers to ethanol- induced hepatic damage. Lancet 1982;2(8310):1243-1245. [ Links ]
66. Mast EE, Margolis HS, Fiore AE, et al. A comprehensive immunisation strategy to eliminate transmission of hepatitis B virus infection in the United States: Recommendations of the Advisory Committee on Immunisation Practices (ACIP) part 1: immunisation of infants, children and adolescents. MMWR Recomm Rep 2005;54(RR-16):1-31. [ Links ]
67. Coffin CS, Terrault NA. Management of hepatitis B in liver transplant recipients. J Viral Hepat 2007;14:37-44. [http://dx.doi.org/10.1111/j.1365-2893.2007.00916.x] [ Links ]
68. Grellier L, Mutimer D, Ahmed M, Brown D, Burroughs AK, Rolles K, et al. Lamivudine prophylaxis against reinfection in liver transplantation for hepatitis B cirrhosis. Lancet 1996;348(9036):1212-1215.[http://dx.doi.org/10.1016/S0140-6736(96)04444-3] [ Links ]
69. Samuel D. Management of hepatitis B in liver transplantation patients. Semin Liver Dis 2004;24:5562.[http://dx.doi.org/10.1055/s-2004-828679] [ Links ]
70. Schiff E, Lai CL, Hadziyannis S, et al. Adefovir dipivoxil for wait-listed and post-liver transplantation patients with lamivudine-resistant hepatitis B: Final long-term results. Liver Transplant 2007;13(3):349-360. [http://dx.doi.org/10.1002/lt.20981] [ Links ]
71. Wachs ME, Amend WJ, Ascher NL, et al The risk of transmission of hepatitis B from HB sAg (-), HB cAb(+), HB IgM(-) organ donors. Transplantation 1995;59(2):230-234. [http://dx.doi.org/10.1097/00007890-199501270-00014] [ Links ]
72. Dickson RC, Everhart JE, Lake JR, et al. Transmission of Hepatitis B by transplantation of livers from donors positive for antibody to hepatitis B core antigen. The National Institute of Diabetes and Digestive and Kidney Diseases Liver Transplantation Database. Gastroenterology 1997;113(5):1668-1674. [http://dx.doi.org/10.1053/gast.1997.v113.pm9352871] [ Links ]
73. Prieto M, Gomez MD, Berenguer M, et al. De novo Hepatitis B after liver transplantation from hepatitis B core antibody-positive donors in an area with high prevalence of anti-HBc positivity in the donor population. Liver Transpl 2001;7(1):51-58.[http://dx.doi.org/10.1053/jlts.2001.20786] [ Links ]
74. Terrault N, Roche B, Samuel D. Management of the Hepatitis B virus in the liver transplantation setting: A European and an American perspective. Liver Transplan 2005;11(7):716-732. [http://dx.doi.org/10.1002/lt.20492] [ Links ]
75. Mutimer D. Review article: Hepatitis B and liver transplantation. Aliment Pharmacol Ther 2006;23(8):1031-1041. [http://dx.doi.org/10.1111/j.1365-2036.2006.02855.x] [ Links ]
76. Bruix J, Sherman M. AASLD Practice Guideline: Management of Hepatocellular Carcinoma: An Update. Hepatology 2011;53(3):1020-1035. [http://dx.doi.org/10.1002/hep.24199] [ Links ]
 Correspondence: C WN Spearman (wendy.spearman@uct.ac.za) and M W Sonderup (msonderup@samedical.co.za)
Correspondence: C WN Spearman (wendy.spearman@uct.ac.za) and M W Sonderup (msonderup@samedical.co.za)


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}