










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSAMJ: South African Medical Journal
On-line version ISSN 2078-5135
Print version ISSN 0256-9574
SAMJ, S. Afr. med. j. vol.102 n.11 Pretoria Jan. 2012
RESEARCH
Lessons from the hepatoblastoma-familial polyposis connection
S W MooreI; N TshifularoII; J J GrobbelaarIII
IMB ChB, FRCS (ED), MD. Division of Paediatric Surgery, Faculty of Medicine and Health Sciences, Stellenbosch University, Tygerberg, Cape Town
IIMB ChB, FCS (SA), FCPaed Surg (SA). Division of Paediatric Surgery, Faculty of Medicine and Health Sciences, Stellenbosch University, Tygerberg, Cape Town
IIIMSc (Genetics). Pathology Research Laboratory and National Health Laboratory Service, Stellenbosch University, Tygerberg, Cape Town
ABSTRACT
BACKGROUND: Approximately one-third of hepatoblastoma (HB) patients have associated congenital abnormalities, but familial recurrence is rare, except in association with familial adenomatous polyposis (FAP). This correlation may be missed if not actively sought, with implications for long-term outcome and management.
METHODS: We retrospectively investigated 3 families with an HB-familial polyposis connection, from a cohort of 113 FAP families (1989 - 2010). Data were analysed to assess clinical problem, treatment, complications and management. Long-term morbidity and functional outcome were analysed to identify management difficulties.
RESULTS: Three FAP families (2.65%) had an HB association. In one case, undiagnosed FAP at the time of HB diagnosis was only detected 5 years later, when the mother presented with advanced colorectal carcinoma. A chromosome 5 APC gene mutation (exon 15 codon 793 C--T) was then identified. In a second case, a non-related boy presented with a stage 4 multifocal HB with lung metastases. Genetic studies identified an APC gene mutation (exon 6 codon 232 C--T). Further family investigation showed >20 related FAP patients. A third HB-FAP association was identified in a known FAP family early in the study, prior to the availability of genetic testing.
CONCLUSION: Although a rare association, a family history of FAP in HB patients is an important 'hidden connection'. Germline variation may be outside the usual FAP gene site. Identifying families with unknown HB/FAP is important due to long-term management implications and follow-up.
Hepatoblastoma (HB) is the most common primary liver cancer of childhood, accounting for up to 1% of all paediatric malignancies, particularly in the younger child. HB is associated with congenital abnormalities in approximately one-third of patients, suggesting complex genetic and/or epigenetic factors in its pathogenesis.1,2
In addition to an association with low birth weight, there are several linked genetic diseases including overgrowth syndromes such as Beckwith-Wiedemann syndrome, chromosomally linked conditions (trisomies 2, 8 and 20) and X-linked Simpson-Golabi-Behmel syndrome, type 1 glycogen storage diseases, Li-Fraumeni syndrome, familial adenomatous polyposis (FAP)3-6 and type 1 neurofibromatosis.7
Familial recurrence of HB is extremely rare outside of associated adenomatous polyposis coli (APC) families,8 and a causative relationship between HB and interstitial deletions of 5q21.3-q23.3 (the APC gene region) is well known.9 Consequently, offspring of FAP families have a 750 - 7 500 times higher risk of developing HB.10,11 Although 75 - 80% of FAP individuals have APC gene mutations, there is a group with non-typical de novo genetic variation.6 As a result, the screening of HB patients for APC gene variation in cases of childhood HB without a family FAP history remains an open discussion. It appears to be important to identify these high-risk individuals and perform long-term screening to improve their management.
We aimed to investigate HB-FAP gene associations and clinical implications in a South African population.
Methods
Based on available clinical data of 2 known FAP families, a local database of 113 FAP cases (1989 - 2010) was investigated for HB associations. Data were analysed for details of clinical problem, treatment, complications and management. Long-term morbidity and functional outcome were analysed to identify management difficulties.
Results
HB was evident in the offspring of 3/113 known FAP cases (2.65%).
Case 1
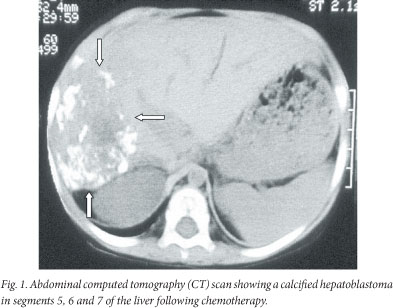
A 2-year-old child re-presented after absconding half-way through a course of chemotherapy (cisplatinum and doxorubicin (PLADO)) for a stage 4 HB. Lung metastases were excised and a right hemi-hepatectomy of a calcified tumour was performed with removal of an additional localised lesion in the left lobe of the liver (Fig. 1). He has survived into adulthood (22 years) without further HB metastatic events. He presented with rectal bleeding at 14 years of age and a pedunculated adenomatous polyp was identified on colonoscopy and removed. He subsequently developed multiple rectal and colonic polyps, for which a total colectomy was performed. Recurrence of rectal polyps in his rectal stump necessitated revision of the lower rectal stump and fashioning of a pouch. Genetic studies identified an APC gene mutation (exon 6 codon 232 C--T). Further investigation of family history revealed relations to a large well-known FAP mixed-ancestry family with >20 affected patients.
Case 2
A mixed-ancestry male infant in a family without any FAP history was diagnosed with HB in the left liver lobe at 15 months of age. Following 4 courses of PLADO, he underwent a left hepatectomy with successful outcome. Six years later, the boy's 34-year-old mother was diagnosed with advanced colon carcinoma and liver metastases. At surgery, multiple colonic polyps were also noted. The mother and child's APC genes showed the same mutation (exon 15 codon 793 C--T). The patient developed multiple adenomatous polyps, and was successfully treated 15 years later by total colectomy. Two other family members with the same genetic mutation have since been identified and followed up.
Case 3
A male infant, born to a known FAP family, was diagnosed and successfully treated for HB early in the study, prior to the availability of genetic testing. The patient never developed further features of FAP and there was no familial recurrence.
Discussion
HB is a well-known embryonal tumour, with recognised genetic associations to several known cancer predisposition syndromes including FAP. Family histories are uncommon in these, with the exception of familial FAP,8 as shown in this study. The incidence of FAP in association with HB, identified in 3/113 FAP families (2.65%), was lower than a previous report of 8/93 (8.6%) HB families.12
There are several lessons from this study. Firstly, although the genetic links between HB and FAP are well established, it is a reminder to clinicians of this rare, possibly hidden link. Secondly, the clinical identification of associated FAP in our first case benefitted the patient and led to the identification of a whole family at high risk of cancer. Thirdly, from the second case, a full history in patients with HB should include a family history of FAP. A timely genetic analysis for FAP in the child with HB would have benefitted the mother who developed an unsuspected metastatic carcinoma colon 6 years later on the basis of unsuspected FAP. Fourthly, predicting HB susceptibility on the basis of the site of APC mutation has proven difficult, as no significant correlation between the site of mutation detected in those with or without HB has been demonstrated.13 The nature of the HB-associated APC mutations, however, appears variable, as shown in 2 families in this study where the APC mutation sites lay outside the loss-of-heterozygosity (LOH)-associated region for colorectal FAP (i.e. codons 1 285 - 1 378),12,14 in keeping with previous reports.8,15
From a molecular perspective, these genetic associations may be a more important link than previously thought in understanding the oncogenesis of liver tumours such as HB. Chromosomal variations occur frequently in HB, having been reported in up to 88% of cases in a genome profiling study.16 The most likely candidate genes identified thus far are CTNNB1 (catenin, beta-1-catenin)17 and insulin-like growth factor II (IGF2) tumour suppressor at locus 11p15.18,19 In addition, more than 85% of HBs show accumulation of β-catenin which indicates an activated Wingless-type (Wnt) pathway.20 Beta-catenin mutations that play a key role in liver development, regeneration and oncogenesis, are found in 50 - 90% of HB tumours.21 However, multiple other deletion or point mutations and frequent gains in chromosomes 1q, 2 (or 2q), 8, 17q, and 20 have been described in HB, as well as losses in chromosomes 4q and 11q, and high-grade amplifications at 7q34, 14q11.2, and 11q22.2.4The HB-related Beckwith-Wiedemann syndrome is also associated with the dysregulation and LOH of imprinted genes at chromosome 11p15.5.22 This subgroup is of considerable interest due to the location of the IGF2 and H19 genes within this region,16 indicating complex genetic/epigenetic associations of the imprinted 11p15 region in the pathogenesis of HB and other related tumours.23,24
The link to FAP is important because of the high risk of developing HB in families with germline APC gene abnormalities. This risk is 750 -7 500 times higher than in the general population,10,11 as well as the risk of other tumours occurring in familial gene carriers. The question as to whether specific APC gene mutations are more likely to be associated with HB remains open. In both cases tested genetically in this series, the APC gene mutation lay outside the usual FAP site on chromosome 5 (viz. family 1, part of a 20-strong known FAP ancestry: exon 6 codon 232 C--T; family 2: exon 15 codon 793 C--T). The significance of this is unknown, but it suggests that those associated with HB may relate to other sites on the gene.
Understanding the link between FAP and HB may, therefore, be of the utmost importance to patient evaluation and follow-up. Screening for APC gene mutations in infants with HB, although difficult, facilitates genetic counselling and an estimation of risk, owing to an autosomal dominant hereditary pattern.11 We agree that the presence of HB-risk APC mutations justifies HB screening in neonates born to gene carriers.12
References
1. Chan PC, Cesta MF, Sills RC, et al. NTP technical report on the toxicology and carcinogenesis studies of beta-myrcene (CAS No. 123-35-3) in F344/N rats and B6C3F1 mice (Gavage studies). Natl Toxicol Program Tech Rep Ser 2010;(557):1-163. [ Links ]
2. Salvatore M, Lorenzetti S, Maranghi F, et al. Molecular link(s) between hepatoblastoma pathogenesis and exposure to di-(2-ethylhexyl) phthalate: a hypothesis. Folia Med (Plovdiv) 2008;50(4):17-23. [ Links ]
3. Li X, Zhao X. Epigenetic regulation of mammalian stem cells. Stem Cells Dev 2008;17(6):1043-1052. [http://dx.doi.org/10.1089/scd.2008.0036] [ Links ]
4. Parada LA, Limon J, Iliszko M, et al. Cytogenetics of hepatoblastoma: Further characterization of 1q rearrangements by fluorescence in situ hybridization. Med Pediatr Oncol 2000;34(3):165-170. [ Links ]
5. Feusner J, Plaschkes J. Hepatoblastoma and low birth weight: a trend or chance observation? Med Pediatr Oncol 2002;39(5):508-509. [http://dx.doi.org/10.1002/mpo.10176; [ Links ]
6. Evers C, Gaspar H, Kloor M, et al. Hepatoblastoma in two siblings and familial adenomatous polyposis: causal nexus or coincidence? Fam Cancer 2012 (in press). [http://dx.doi.org/10.1007/s10689-012-9538-2; [ Links ]
7. Ucar C, Caliskan U, Toy H, Gunel E. Hepatoblastoma in a child with neurofibromatosis type I. Pediatr Blood Cancer 2007;49(3):357-359. [http://dx.doi.org/10.1002/pbc.20663] [ Links ]
8. Giardiello FM, Petersen GM, Brensinger JD, et al. Hepatoblastoma and APC gene mutation in familial adenomatous polyposis. Gut 1996;39(6):867-869. [ Links ]
9. Half E, Bercovich D, Rozen P. Familial adenomatous polyposis. Orphanet J Rare Dis 2009;4:22. [http://dx.doi.org/10.1186/1750-1172-4-22] [ Links ]
10. Hughes LJ, Michels VV. Risk of hepatoblastoma in familial adenomatous polyposis. Am J Med Genet 1992;43(6):1023-1025. [http://dx.doi.org/10.1002/ajmg.1320430621] [ Links ]
11. Aretz S, Koch A, Uhlhaas S, et al. Should children at risk for familial adenomatous polyposis be screened for hepatoblastoma and children with apparently sporadic hepatoblastoma be screened for APC germline mutations? Pediatr Blood Cancer 2006;47(6):811-818. [http://dx.doi.org/10.1002/pbc.20698] [ Links ]
12. Crabtree M, Sieber OM, Lipton L, et al. Refining the relation between 'first hits' and 'second hits' at the APC locus: the loose fit' model and evidence for differences in somatic mutation spectra among patients. Oncogene 2003;22(27):4257-4265. [http://dx.doi.org/10.1038/sj.onc.1206471] [ Links ]
13. Hirschman BA, Pollock BH, Tomlinson GE. The spectrum of APC mutations in children with hepatoblastoma from familial adenomatous polyposis kindreds. J Pediatr 2005;147(2):263-266. [http://dx.doi.org/10.1016/j.jpeds.2005.04.019] [ Links ]
14. Cetta F, Montalto G, Gori M, et al. Germline mutations of the APC gene in patients with familial adenomatous polyposis-associated thyroid carcinoma: Results from a European cooperative study. J Clin Endocrinol Metab 2000;85(1):286-292. [ Links ]
15. Suzuki M, Kato M, Yuyan C, et al. Whole-genome profiling of chromosomal aberrations in hepatoblastoma using high-density single-nucleotide polymorphism genotyping microarrays. Cancer Sci 2008;99(3):564-570. [http://dx.doi.org/10.1111/j.1349-7006.2007.00710.x] [ Links ]
16. Blaker H, Hofmann WJ, Rieker RJ, et al. Beta-catenin accumulation and mutation of the CTNNB1 gene in hepatoblastoma. Genes Chromosomes Cancer 1999;25(4):399-402. [ Links ]
17. Jeng YM, Wu MZ, Mao TL, et al. Somatic mutations of beta-catenin play a crucial role in the tumorigenesis of sporadic hepatoblastoma. Cancer Lett 2000;152(1):45-51. [ Links ]
18. Chitragar S, Iyer VK, Agarwala S, et al. Loss of heterozygosity on chromosome 11p15.5 and relapse in hepatoblastomas. Eur J Pediatr Surg 2011;21(1):50-53. [http://dx.doi.org/10.1055/s-0030-1267208] [ Links ]
19. Wirths O, Waha A, Weggen S, et al. Overexpression of human Dickkopf-1, an antagonist of wingless/ WNT signaling, in human hepatoblastomas and Wilms' tumors. Lab Invest 2003;83(3):429-434. [ Links ]
20. Armengol C, Cairo S, Fabre M, Buendia MA. Wnt signaling and hepatocarcinogenesis: the hepatoblastoma model. Int J Biochem Cell Biol 2011;43(2):265-270. [http://dx.doi.org/10.1016/j.bioceL2009.07.012] [ Links ]
21. De Crescenzo A, Coppola F, Falco P, et al. A novel microdeletion in the IGF2/H19 imprinting centre region defines a recurrent mutation mechanism in familial Beckwith-Wiedemann syndrome. Eur J Med Genet 2011;54(4) e451-e454. [http://dx.doi.org/10.1016/j.ejmg.2011.04.009] [ Links ]
22. Smith AC, Squire JA, Thorner P, et al. Association of alveolar rhabdomyosarcoma with Beckwith-Wiedemann syndrome. Pediatr Dev Pathol 2001;4:550-558. [ Links ]
23. Weksberg R, Shuman C, Beckwith JB. Beckwith-Wiedemann syndrome. Eur J Hum Genet 2009;18(1):8-14. [http://dx.doi.org/10.1038/ejhg.2009.106] [ Links ]
24. Weksberg R, Shuman C, Beckwith JB. Beckwith-Wiedemann syndrome. Eur J Hum Genet 2009; 18(1):8-14. [ Links ]
Accepted 7 August 2012.
 Corresponding author:
Corresponding author:
S W Moore
(swm@sun.ac.za)

