










Serviços Personalizados
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkSAMJ: South African Medical Journal
versão On-line ISSN 2078-5135
versão impressa ISSN 0256-9574
SAMJ, S. Afr. med. j. vol.102 no.9 Pretoria Set. 2012
RESEARCH
The burden of sickle cell disease in Cape Town
Ambroise WonkamI; Chido PondeIV; Nan NicholsonV; Karen FieggenII; Raj RamesarIII; Alan DavidsonVI
IMD, Dip Medical Genetics. Division of Human Genetics, Department of Clinical Laboratory Sciences, Faculty of Health Sciences, University of Cape Town
IIMB ChB, FCPaed, Cert Clinical Genetics. Division of Human Genetics, Department of Clinical Laboratory Sciences, Faculty of Health Sciences, University of Cape Town
IIIPhD. Division of Human Genetics, Department of Clinical Laboratory Sciences, Faculty of Health Sciences, University of Cape Town
IVMedical student. Faculty of Health Sciences, University of Cape Town
VMB ChB. Haematology/Oncology Service, Red Cross War Memorial Children's Hospital, and Department of Paediatrics and Child Health, University of Cape Town
VIMB ChB, FCPaed (SA). Haematology/Oncology Service, Red Cross War Memorial Children's Hospital, and Department of Paediatrics and Child Health, University of Cape Town
ABSTRACT
BACKGROUND: South Africa has a low incidence of sickle cell disease (SCD). However, its demographics are changing because of immigration from sub-Saharan African countries where SCD is prevalent.
OBJECTIVES: We aimed to determine the frequency of SCD presenting to the Haematology/Oncology Service at Red Cross War Memorial Children's Hospital in Cape Town and to measure the associated disease burden.
METHODS: This was a retrospective cross-sectional study of patients first attending the Haematology Service between January 2001 and June 2010.
RESULTS: A total of 58 SCD patients were indentified, with an annual frequency that increased over the study period by 300 -400%. Up to 93.1% (n=54) were originally from other African countries, mainly the Democratic Republic of Congo (62.1%, n=36). One patient had sickle D-Punjab genotype, and all the other patients had the homozygous sickle cell anaemia genotype (Hb SS). Their haematological parameters demonstrated a normocytic anaemia with high white cell counts. The mean number of clinic visits per patient per year was 22.2 (range 0 - 64), and the mean number of hospital admissions per patient per year was 1.2 (range 0 - 5). All the patients were on antibiotic prophylaxis. The majority had at least one blood transfusion (65.5%, n=38), and a significant proportion required intravenous analgesia on admission (29.3%, n=17) and hydroxyurea treatment (36.2%, n=21).
CONCLUSIONS: Over the past 10 years the frequency of SCD has increased considerably, imposing a significant burden and new challenges to the health services in Cape Town.
Sickle cell disease (SCD), the commonest monogenic disease in humans, is caused by a point mutation leading to a single amino acid substitution in the beta-subunit of haemoglobin (Hb), the principal oxygen transporter in red blood cells. In sub-Saharan Africa (SSA), SCD occurs at its highest frequency, and the prevalence of sickle cell trait is estimated to be between 5% and 40%, with up to 300 000 affected babies born each year.1 A notable feature of SCD is the frequent occurrence of acute episodes of pain, which are generally attributable to vaso-occlusive crises.2-4 As a consequence of these complications, SCD patients have increased mortality compared with control populations.5 The health care costs of people with SCD are substantial, as the disease is chronic. Individuals with SCD will require lifelong prophylactic penicillin and folate and in some cases hydroxyurea, which acts by increasing the concentration of fetal haemoglobin (HbF) and reduces painful episodes, acute chest syndrome and the need for blood transfusions.6,7 It has been estimated that SCD results in the annual loss of several millions of disability-adjusted life years, particularly in the developing world.2 Haemoglobinopathies alone represent a health/disease burden comparative to that of communicable and other major diseases.1
The incidence of SCD in South Africa is very low,8 but the ethnic diversity in South Africa means that its prevalence differs for each ethnic group. Several haemoglobin variants occur in mixed-ancestry populations in South Africa,9 the most common being HbE and HbS. HbE probably originated from South-East Asia and HbS from East Africa and possibly Madagascar.10 The sickle cell gene is found in the South African Indian population,11 and cases of sickle cell-C haemoglobin disease and sickle cell-beta thalassaemia have occurred in white South African families who are not of Mediterranean descent.12 The heterozygous trait can be found in the indigenous Venda and Shangaan South African ethnic groups; these are two of the least numerous ethnic groups and the trait is found at a low frequency of approximately 0.2%, making SCD very uncommon among South African blacks.13
The demographics of South Africa are changing because of the influx of immigrants from other parts of Africa, particularly Central Africa, where SCD is prevalent. This is likely to change the frequency of SCD and the associated disease burden in this country.
The Haematology/Oncology Service at Red Cross War Memorial Children's Hospital (RCH) in Cape Town, a tertiary paediatric hospital, currently sees about 900 outpatients with complex haematological disorders each year, and admits about 250 of these patients annually. With the increasing numbers of SCD patients, the service has developed SCD-specific treatment guidelines and since 2009 has sponsored the development of satellite clinics at New Somerset Hospital and Victoria Hospital, two secondary-level hospitals in the Cape Town metropolitan area. Stable patients were initially referred to these clinics, but they now enroll a growing number of patients independently. Unless patients develop complications they are not seen at RCH.
We aimed to determine the frequency of SCD and the associated disease burden seen at RCH.
Methods
This was a 10-year retrospective cross-sectional study of the hospital attendances of all SCD patients at RCH, from January 2001 to June 2010. Data collection took 2 weeks. The study was performed as a Short Study Module, which aims to introduce University of Cape Town medical students to research, and was approved by the University of Cape Town, Faculty of Health Sciences Ethics Committee.
The data sheet consisted of socio-demographic data, hospital attendances and admissions, clinical events and interventions, and the haematological indices of the SCD patients. The data were collected from the patients' files and when possible by phone interviews with caregivers to complete missing socio-demographic data.
Descriptive statistics were used to analyse the data.
Results
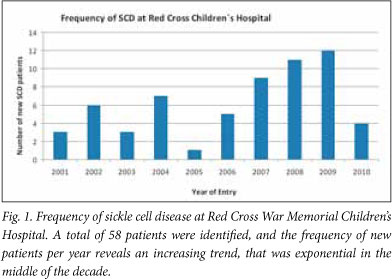
A total of 58 patients were identified. The frequency of new patients per year reveals an increasing trend over the past 10 years, with an increase of 300 - 400% over the study period (Fig. 1).
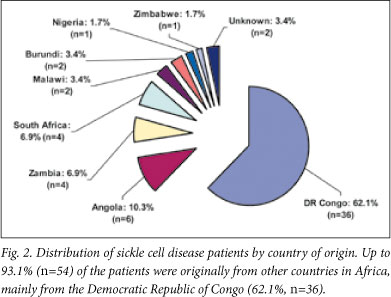
Up to 93.1% (n=54) of the patients were originally from other countries in Africa, mainly the Democratic Republic of Congo (DRC) (62.1%, n=36) (Fig. 2). The mean age at first attendance was 8 years (range 2 - 21 years).
A total of 22.4% (n=13) of families had at least one other SCD-affected sibling, and in 3 families (5.2%) at least one other child had died due to SCD.
Up to 41.2 % of the fathers and 37.5% of the mothers of the patients were unemployed.
High-performance liquid chromatography (HPLC) haemoglobin electrophoresis revealed that all patients had the sickle cell anaemia genotype (Hb SS), except for one patient who had the sickle D-Punjab genotype.
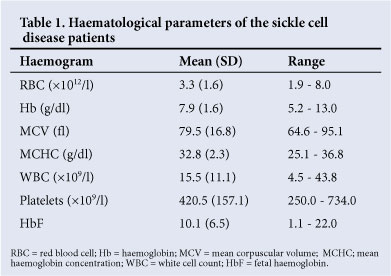
Routine full blood counts showed that, on average, the SCD patients had a normocytic, normochromic anaemia with a high white cell count, the latter usually indicative of excess normoblasts in the peripheral blood. The mean HbF level was relatively high at 10.1% (Table 1).
All the patients had regular follow-up. The mean number of clinic visits per patient per year was 22.2 (range 0 - 64); 70.7% (n=41) of patients had been admitted at least once, and the mean number of hospital admissions was 1.2 per patients per year (range 0 - 5). Only 37.9% (n=22) had at least one painful crisis requiring hospital admission; the mean number of painful crises per patient per year requiring hospital admission was 0.7 (range 0 - 3) (Table 2).
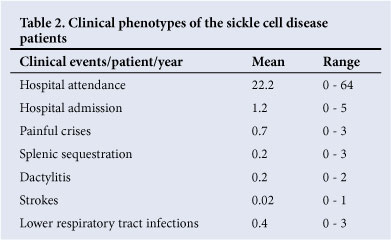
One patient (1.7%) was reported to have suffered a stroke, which was confirmed on a computed tomography scan. Five (8.6%) had acute splenic sequestration and 21 (36.2%) suffered lower respiratory tract infections.
All the patients were maintained on antibiotic prophylaxis and folic acid, 69% (n=40) had received oral analgesia and 66.5% (n=38) blood transfusion, and 29.3% (n=17) had received intravenous analgesia on admission and 36.2% (n=21) hydroxyurea treatment.
There were no deaths recorded in this cohort of SCD patients.
In 2010 - 2011, 4 parents underwent prenatal genetic testing for SCD that had recently been introduced by the National Health Laboratory Service laboratory of the Division of Human Genetics at UCT. All were citizens of the DRC and had an SCD-affected child. Restriction fragment length polymorphism-polymerase chain reaction (RFLP-PCR) diagnosed 1 homozygous normal fetus (HbAA), 2 heterozygous pregnancies (HbAS), and 1 fetus with SCD. The parents of the SCD-affected fetus opted for termination of the pregnancy.
Discussion
We believe that this study represents the first quantitative evidence of the increase in SCD in a hospital setting in South Africa over the past decade. We suspect that this trend would be similar in many hospitals serving migrant populations from SSA, and it has been noted in Northern Europe, where the number of patients with SCD is steadily increasing as a result of migration.14 Unless SSA countries reach more political and economic stability, and achieve better growth, this trend could continue.
This increasing number of SCD patients has spurred the development of disease-specific guidelines at RCH and the establishment of satellite clinics at two secondary-level hospitals. Moreover, our data are an underestimation of SCD patients in Cape Town, because some patients are now diagnosed and followed up entirely at secondary-level hospitals.
SCD imposes a significant burden on health resources. Patients require regular monitoring, as evidenced by their high number of hospital attendances and admissions, and there is a need for blood transfusion and/or the administration of intravenous analgesia. Those treated with hydroxyurea also require careful assessment and follow-up. The average number of painful episodes requiring hospitalisation (>3 per year), strokes (>1 per year) or acute chest syndrome presentations (>3 per year), and deaths due to SCD (>1 in the family), have been used as proxies of severity in SCD families in the Cooperative Study of SCD in the USA.3-5 Rates of these clinical events were relatively low in our sample (Table 2), reflecting the possibility of a moderate phenotype. A favourable medical setting in South Africa and a malaria-free environment could have also contributed to a milder phenotype. In addition, the moderate phenotype could also be attributed to the use of hydroxyurea in 36% of patients, since this treatment has been associated with a mild SCD phenotype.7 The use of hydroxyurea could also explain the high mean HbF level (10.1%) in our cohort. Studies in Saudi Arabia, where the Asian β-globin haplotype predominates and SCD patients have HbF levels of 10 - 40%, report that patients rarely have severe disease.15 In Tanzania, the mean HbF level in 1 668 SCD patients was 6.3%,16 comparable to 7% in the USA5 but much lower than 27.8% in Arab countries.15 Nevertheless, haematological data for our sample should be interpreted with caution, as this was a retrospective study based on information from the medical folder of a tertiary hospital. Haematological indices presented here are those documented at first visit to RCH. Unfortunately prior therapy with transfusion and or hydroxyurea was not always noted. In addition, the frequent use of hydroxyurea in this group of SCD patients (36%) could be explained by an increased disease severity in our population. In our service two secondary-level hospitals, namely New Somerset Hospital and Victoria Hospital, manage some patients independently, referring only those with complications to RCH.
The fact that only one stroke was recorded is notable, since primary prophylaxis with chronic blood transfusion cannot be offered to those at risk because of inability to monitor patients for abnormal transcranial Doppler abnormalities. This strategy is known to reduce stroke episodes.17
The absence of any SCD-related mortality must be interpreted with caution. A retrospective cross-sectional hospital-based study may miss children with severe disease who have died in early childhood, and there may be individuals who have a mild disease phenotype and are never diagnosed.18 In addition some patients may have died in secondary hospitals, though we are not aware of any such cases.
The direct costs of the care of SCD at RCH requires quantitative evaluation to plan future services for the care and prevention of SCD in Cape Town, which could also be applicable at other institutions caring for SCD in South Africa. The burden of SCD on the affected families should also be considered. There are direct (medical costs and transport) and indirect costs (loss of employment) that put family finances under pressure, which is exacerbated by the fact that many of the parents are not employed. In addition, patients and their families suffer psychosocial stresses. Our growing experience with prenatal diagnosis demonstrates that while few of our parents have received genetic counselling, those who were candidates all accepted prenatal testing, and the family with a SCD-affected fetus elected to terminate the pregnancy.
It was anticipated more than a decade ago that the multicultural nature of the South African nation, with migration from SSA, would lead to the introduction of the HbS gene into South African populations that did not have it.19 The small population of 'indigenous' South Africans suggests a low prevalence of SCD. We have not explored the origin of SCD mutations in these patients.
As long as migration of people from other African countries continues, and improved services lead to the increased survival of individuals with SCD to reproductive age, the frequency and prevalence of SCD will continue to increase in South Africa. Migration and its impact on disease burden is a sensitive issue globally, and specifically in multicultural South Africa. However, we need to address the resulting challenges to improve care and prevention of SCD. We hope that our basic statistics support this reality and open discussions in a way that avoids unfair stigmatisation. National and international agencies involved in the care and prevention of SCD need to be aware of the full extent of the problem in SSA, where the disease is traditionally prevalent, but also in countries like South Africa where migration is changing the frequency of SCD in tertiary hospitals such as RCH in Cape Town.
Acknowledgements. We thank the staff of the Haematology/Oncology Service of Red Cross War Memorial Children's Hospital, and the patients and parents for their participation.
Conflict of interest. No conflict of interest to declare.
References
1. Weatherall DJ. Hemoglobinopathies worldwide: present and future. Curr Mol Med 2008;8(7):592-599. [ Links ]
2. Weatherall DJ. The inherited diseases of hemoglobin are an emerging global health burden. Blood 2010;115(22):4331-4336. [ Links ]
3. Malowany JI, Butany J. Pathology of sickle cell disease. Semin Diagn Pathol 2012;29(1):49-55. [ Links ]
4. Bartolucci P, Galactéros F. Clinical management of adult sickle-cell disease. Curr Opin Hematol 2012;19(3):149-155. [ Links ]
5. Platt OS, Brambilla DJ, Rosse WF, et al. Mortality in sickle cell disease: Life expectancy and risk factors for early death. N Engl J Med 1994;330:1639-1644. [ Links ]
6. Lanzkron S, Strouse JJ, Wilson R, et al. Systematic review: Hydroxyurea for the treatment of adults with sickle cell disease. Ann Intern Med 2008;148(12):939-955. [ Links ]
7. Strouse JJ, Lanzkron S, Beach MC, et al. Hydroxyurea for sickle cell disease: a systematic review for efficacy and toxicity in children. Pediatrics 2008;122(6):1332-1342. [ Links ]
8. Bonafede RP, Botha MC, Beighton P. Inherited anaemias in the Greek community of Cape Town. J Med Genet 1979;16(3):197-200. [ Links ]
9. Bird AR, Ellis P, Wood K, Mathew C, Karabus C. Inherited haemoglobin variants in a South African population. J Med Genet 1987;24(4):215-219. [ Links ]
10. Bird AR, Karabus CD, Hartley PS. Microcytic anaemia and haemoglobinopathy in Cape Town children. S Afr Med J 1982;62(13):429-430. [ Links ]
11. Reddy MV, Ward FA .The sickle-cell phenomenon in South Africa. S Afr Med J 1969;43:1217-1219. [ Links ]
12. Dunston T, Rowland R, Huntsman RG, Yawson G Sickle-cell haemoglobin C disease and sickle-cell beta thalassemia in white South Africans. S Afr Med J 1972;46:1423-1426. [ Links ]
13. Beighton P, Botha MC.Inherited disorders in the black population of southern Africa. Part I. Historical and demographic background; genetic haematological conditions. S Afr Med J 1986;69(4):247-249. [ Links ]
14. Howard J, Davies SC. Sickle cell disease in Northern Europe. Scandinavian Journal of Clinical Laboratory Investigation 2007;67(1):27-38. [ Links ]
15. Labie D, Pagnier J, Lapoumeroulie C, et al. Common haplotype dependency of high G gamma-globin gene expression and high Hb F levels in beta-thalassemia and sickle cell anemia patients. Proceedings of the National Academy of Sciences (PNAS) 1985;82:2111-2114. [ Links ]
16. Makani J, Menzel S, Nkya S, et al. Genetics of fetal hemoglobin in Tanzanian and British patients with sickle cell anemia. Blood 2011;117(4):1390-1392. [ Links ]
17. Aygun B, Wruck LM, Schultz WH, et al. Chronic transfusion practices for prevention of primary stroke in children with sickle cell anemia and abnormal TCD velocities. Am J Hematol 2012;87(4):428-430. [ Links ]
18. Makani J, Cox SE, Soka D, et al. Mortality in sickle cell anaemia in Africa: A prospective cohort study in Tanzania. PLoS One 2011;6(2):e14699. [ Links ]
19. Aluoch J, Jacobs P. Sickle-cell disease in sub-Saharan Africa - past, present and future status. S Afr Med J 1996;86(8):982-983. [ Links ]
Accepted 4 June 2012.
Corresponding author: A Wonkam (ambroise.wonkam@uct.ac.za)

