










Serviços Personalizados
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkSAMJ: South African Medical Journal
versão On-line ISSN 2078-5135
versão impressa ISSN 0256-9574
SAMJ, S. Afr. med. j. vol.102 no.8 Pretoria Ago. 2012
GUIDELINES
South African guidelines for the management of Gaucher disease, 2011
Lysosomal Storage Disorder Medical Advisory Board (in alphabetical order); Louisa Bhengu; Alan Davidson; Paul du Toit; Trevor Gerntholtz; Kenny Govendrageloo; Rene Heitner; Bertram Henderson; Lawrence Mubaiwa; Sheeba Varughese
ABSTRACT
BACKGROUND: Gaucher disease is an autosomal recessive lysosomal glycosphingolipid storage disorder resulting from a deficiency of lysosomal enzyme acid â-glucosidase (glucocerebrosidase). This partial enzyme deficiency results in accumulation of glycosphingolipid-laden macrophages (Gaucher cells) throughout the liver, spleen, bone marrow, skeleton, lungs and brain (only in types 2 and 3).
OBJECTIVE: These guidelines aim to provide a standard of care for patients with Gaucher disease in keeping with international standards, but also realistic for South Africa, and to provide a shared-care model for treating physicians and funders regarding care for these patients.
RECOMMENDATIONS: All healthcare professionals involved in the diagnosis and management of Gaucher disease should take note of and implement these guidelines in clinical practice as far as possible.
VALIDATION: These guidelines were developed through consensus by the Lysosomal Storage Disorder Medical Advisory Board. They are largely based on the UK 2005 National Guidelines for Gaucher Disease, but include new treatment recommendations for enzyme replacement therapy based on subsequent publications. The Southern African Society for Human Genetics (SASHG) (who have endorsed the guidelines) and the National Osteoporosis Foundation of South Africa (NOFSA) provided valuable input.
GUIDELINES SPONSOR: Genzyme initiated the project and sponsored the meetings of the Advisory Board and all costs generated by these meetings.
CONCLUSION: It is intended that these guidelines will enable all patients suffering from Gaucher disease to be diagnosed and offered the best possible care available.
1. Background
The South African Lysosomal Storage Disorder (LSD) Medical Advisory Board (MAB) is a multidisciplinary/multispecialty team of physicians from academic and private healthcare institutions with an interest in treating LSDs. The multidisciplinary approach is essential because of the multisystem nature of the disorders. The LSD MAB members are independent and aim to provide a shared-care model for treating physicians and funders regarding various aspects of care for these patients.
The original UK 2005 National Guidelines for Gaucher Disease1,2 are based on limited natural history and literature available at that time. Since these guidelines were written, three important publications have emerged.3-5 They demonstrate a dose response to enzyme replacement therapy of up to 60 U/kg every other week (EOW) on haematological and visceral parameters, a similar dose response on bone mineral density, and a relationship between timing of initiation of therapy and reduced risk of avascular necrosis.1-3
The South African guidelines are based primarily on the UK guidelines. However, the LSD MAB included evidence from subsequent publications to provide an international standard of care, but also realistic for South Africa, given the constraints facing healthcare workers and patients. These guidelines are intended to enable all patients suffering from Gaucher disease to be diagnosed and offered the best possible care available.
Since Gaucher disease is rare, it is difficult to obtain large numbers of patients for studies. The availability of effective treatment makes randomised control studies ethically difficult to design and perform. Thus most of the data are derived from case reports and registries.
2. Disease overview
Gaucher disease is an autosomal recessive lysosomal glycosphingolipid storage disorder resulting from a deficiency of the lysosomal enzyme acid â-glucosidase (glucocerebrosidase).1 It is the most prevalent of the LSDs and is divided into 3 clinical types, of which type 1 is by far the most common.1 This partial enzyme deficiency results in accumulation of glycosphingolipid-laden macrophages (Gaucher cells) throughout the liver, spleen, bone marrow, skeleton and occasionally the lung.1 In types 2 and 3 pathology also occurs within the brain.1 The onset of clinical features may occur at any age in type 1 disease, while type 2 presents in infancy and type 3 typically in early childhood.1 Early onset in type 1 disease predicts a more aggressive course.1 Gaucher disease has been demonstrated to occur in all ethnic groups in South Africa. Some population groups such as the Ashkenazi Jews have a higher incidence.
3. Diagnosis
The diagnosis of Gaucher disease is based on history, clinical evaluation, laboratory investigations and diagnostic imaging.
3.1 Baseline assessments1
History, including family pedigree: medical history of painful hips, bone crises, fractures, orthopaedic interventions; surgical history, including splenectomy; history of respiratory tract infections or other frequent infections; history of eye movement abnormalities, squints; history of bleeding, bruising, blood transfusions, nose bleeds, onset of menarche and/or heavy menstruation; history of hepatosplenomegaly or abdominal distension.
3.2 Clinical assessments1
Height, weight, blood pressure and urine analysis; evidence of corneal opacification should be sought; oculomotor gaze palsies and pingueculae; presence of jaundice, pallor, ecchymoses, purpura and petechiae; spinal deformity, defects of joint movement particularly hip assessment; presence of visceromegaly (hepatosplenomegaly) and signs of chronic liver disease; neurological evaluation to include symptoms of parkinsonism for evaluation of type of Gaucher disease; and quality of life questionnaire.
3.3 Laboratory evaluations1
Baseline enzyme activity of glucocerebrosidase in leukocytes
DNA mutational analysis (at baseline only)
Bone marrow aspiration is not required for diagnosis. Recommended:1
Chitotriosidase activity is elevated in Gaucher disease (except in a small proportion of patients where it is absent due to a mutation that can be identified by DNA mutation analysis), and can be assessed at baseline and as required to monitor disease activity. Other biomarkers which decrease with effective enzyme replacement include serum angiotensin-converting enzyme (ACE) and tartrate-resistant acid phosphatase (TRAP).
Full blood count (FBC) with peripheral smear, vitamin B12, folate, iron and ferritin.
Endocrine profile: parathyroid hormone (PTH) & 25-OH vitamin D & thyroid-stimulating hormone (TSH) (free thyroxine (T4) & free triiodothyronine (T3) when indicated).
Biochemistry profile: renal, liver and lipid profiles, C-reactive protein (CRP), immunoglobulins, serum protein electrophoresis.
Clotting screen: international normalised ratio (INR), prothrombin time (PT), partial thromboplastin time (PTT) (factor XI levels and fibrinogen when necessary, particularly in pregnancy).
Serum storage for antibody screening (store for baseline) with protocol and informed consent.
HIV screening with informed consent; and hepatitis screen.
For timing of tests refer to section 7 (some blood tests are not available in South Africa and can be done in a European laboratory or specimens collected and frozen for later analysis as required).
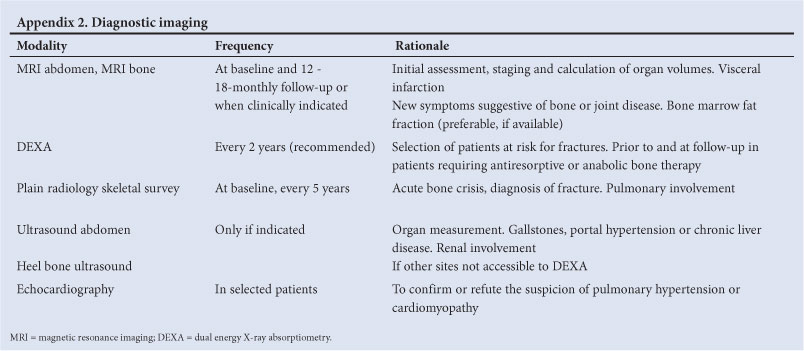
3.4 Diagnostic and follow-up imaging1
3.4.1 Magnetic resonance imaging (MRI): abdomen and high-risk bones (femur, humerus, lumbar vertebrae, hip joints) at baseline, when clinically indicated and at dose adjustments: (i) initial assessment, staging and calculation of organ volumes; (ii) visceral infarction - only if necessary for clinical evaluation; (iii) new symptoms suggestive of bone or joint disease (bone marrow burden score6 is optional). MRI is valuable for assessment of structural bone abnormalities.
3.4.2 Dual energy X-ray absorptiometry (DEXA): at baseline and as needed. Identification of patients at risk of fractures, and prior to and at follow-up of patients requiring antiresorptive or anabolic bone therapy. For paediatric patients, DEXA scan must be performed at a centre that uses paediatric reference values. DEXA measurements should be done at bone sites that are free of osteonecrosis. DEXA is the method of choice for evaluating osteopenia/osteoporosis.
3.4.3 Plain radiology: skeletal survey when clinically indicated for acute bone crisis or diagnosis of fracture; chest X-ray (CXR) for suspected pulmonary involvement.
3.4.4 Ultrasound: organ measurement (liver and spleen size when volumetric MRI not available), gall stones, portal hypertension or chronic liver disease, renal involvement. Heel ultrasound if indicated to assess bone involvement if evaluation at other sites not possible by DEXA.
3.5 Cardiopulmonary evaluation1
CXR, pulmonary function tests and electrocardiogram (ECG) at baseline and as indicated. Echocardiography in selected patients to confirm or refute the suspicion of pulmonary hypertension or cardiomyopathy
4. Treatment
4.1 Criteria for treatment1
In general, presentation with clinical features of Gaucher disease;7 one aim of intervention in such patients is primary prevention of irreversible organ damage, including bone, spleen or liver damage,8 pulmonary9 and neurological complications.1
4.2 When to start - indications for specific treatment1'9
Immediate initiation of treatment:
Clinically significant thrombocytopenia or anaemia
History of progressive organomegaly (hepatosplenomegaly)
Bone involvement
Growth failure/retardation
Delayed puberty
Pulmonary involvement
History of splenectomy
Impairment of function (physical and quality of life)
Any child with disease manifestations should be treated.
4.3 Treatment options
4.3.1 Enzyme replacement therapy (ERT):1 imiglucerase, an analogue of human intracellular glucocerebrosidase, is the treatment of choice for types 1 and 3 Gaucher disease.
4.3.2 Supportive therapy:1 indicated for those patients who decline the above option (usually elderly patients) and require symptomatic supportive intervention with blood products, bisphosphonate therapy, and/or analgesia.
4.3.3 Mobility aids:1 e.g. crutches and wheelchairs to aid mobility for everyday living.
4.3.4 Experimental therapies:1 might become available and will need further evaluation at such time.
4.3.5 Monitoring:1 patients identified with Gaucher disease mutations, who may be asymptomatic, do not require treatment at present, but must be monitored regularly (6-monthly) for disease progression according to the goals of treatment (section 5) and indications to start therapy as above. Treatment options for all patients must be reviewed according to clinical and laboratory data.
4.3.6 Bone marrow transplantation:1 no longer advocated for type 1 patients now that effective ERT is available; may still be considered under certain circumstances for type 3 patients when a matched, unaffected sibling donor has been identified.
4.3.7 Other therapies: substrate reduction therapy is currently utilised in some patients with mild disease. It is currently not available in South Africa.
4.3.8 Genetic counselling:1 this is an important component of supportive care for any family and best provided by a healthcare professional well versed in these aspects of care. Parents of affected individuals, individuals themselves when they reach an age of understanding, siblings of carriers or affected individuals, and spouses/potential spouses should be included. Genetic counselling aims to enable the patients and their families to understand the medical facts, role of inheritance, strategies to prevent recurrence and to make the best possible adaptation to the disorder. Autosomal recessive inheritance implies that both parents of an affected person are carriers of a mutated acid â-glucosidase gene. The affected child has inherited a mutated copy from each parent. The parents have a 25% chance for another affected child in subsequent pregnancies. The affected person must also understand the risk of transmission of a mutated gene to their offspring.
Carrier testing is therefore important for these families. Whereas enzyme activity is a reliable method for the diagnosis of Gaucher disease, its use for identifying carriers is limited. The best method for carrier testing and prenatal diagnosis is mutation analysis.
4.4 Imiglucerase treatment - dosing regimen10
The only product currently registered for Gaucher disease in South Africa is imiglucerase that is administered by intravenous infusion over 1 - 2 hours. Dosage should be individualised to each patient and dosage adjustments made on an individual basis. This may increase or decrease, based on achievement of therapeutic goals as assessed by routine comprehensive evaluations of the patient's clinical manifestations. Dose is also partly determined by the requirement to use whole vials of imiglucerase, which is available in 400 U vials. Therapy may be continued throughout pregnancy at the discretion of the treating physician.11
4.4.1 Dose selection based on severity: demonstration of a dose response to imiglucerase for haematological and visceral parameters3 and bone mineral density1 implies a need and provides a rationale for initial dose selection based on clinical severity, followed by dose titration (up or down) according to achievement of therapeutic goals, and specific symptoms that are considered severe or higher risk. In specific cases, dose adjustments might be required according to clinical need.
Patients fulfilling one of the severe or high-risk criteria should start imiglucerase at a recommended dose of 30 - 60 U/kg every other week (EOW). All other patients, not fulfilling high-risk criteria, but fulfilling the 'indications for specific treatment', should commence imiglucerase at a recommended dose of 10 - 30 U/kg EOW (see 4.4.2).
4.4.2 Severe or high-risk disease (30 - 60 U/kg EOW): one of the following criteria:
Symptomatic paediatric patients identified with severe genotypes known to be associated with rapid disease progression
Paediatric patients prior to epiphyseal closure with evidence of growth failure and/or delayed puberty
The presence of bone disease comprising bone crises, bone pain, pathological fracture, radiological evidence of osteonecrosis, infarction or generalised osteopenia (previous joint replacement will be considered an indication of severe bone disease)
Splenectomy
Significant splenomegaly as indicated by: spleen volume at least 6 times normal OR at risk of splenic rupture OR severe symptoms of abdominal distension or pain OR hypersplenism OR episodes of splenic infarction
Evidence of hepatic infarction or cirrhosis or complications
Transfusion dependency OR coagulopathy OR platelets under 20 000/ì! (20x107l)
Pulmonary involvement
Type 3 Gaucher disease. There are no known drug interactions with imiglucerase.10
Co-morbidities are not an indication to withhold therapy.10 Refer to the South African prescribing information for imiglucerase.
5. Goals of treatment
5.1 Treatment goals for anaemia1,9 (Table 1)

Achieve acceptable levels of haemoglobin (Hb); eliminate blood transfusion dependency; reduce fatigue, dyspnoea, angina; maintain improved Hb values achieved after the first 12 - 24 months of therapy.
Treatment response may vary. Mild anaemia may persist especially in patients with severe splenic enlargement.8
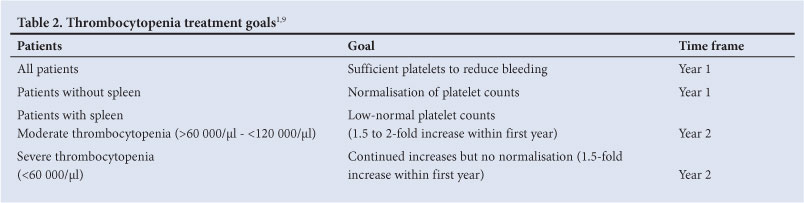
5.2 Treatment goals for thrombocytopenia1,9 (Table 2)
All patients: increase platelet counts during the first year of treatment sufficiently to prevent surgical, obstetric and spontaneous bleeding.
Splenectomised patients: normalisation of platelet count by 1 year of treatment.
Patients with intact spleen:
Moderate baseline thrombocytopenia (60 000 - 120 000/ìÏ: the platelet count should increase by 1.5 to 2-fold by year 1 and approach low-normal levels by year 2
Severe baseline thrombocytopenia (<60 000/ì!): the platelet count should increase by 1.5-fold by year 1 and continue to increase slightly during years 2 - 5 (doubling by year 2), but normalisation is not expected
Avoid splenectomy (may be necessary for life-threatening haemorrhagic events)
Maintain stable platelet counts to eliminate risk of bleeding after a maximal response has been achieved.
Treatment response may vary.9
5.3 Treatment goals for splenomegaly1,9 (Fig. 1)
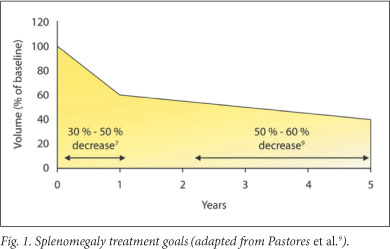
Reduce and maintain spleen volume to <2 - 8 times normal; reduce the spleen volume by 30 - 50% within year 1 and by 50 - 60% by years 2 - 5; alleviate symptoms due to splenomegaly: abdominal distension, early satiety and new splenic infarction; eliminate hypersplenism; avoid splenectomy.
Treatment response may vary. Patients with severe splenic enlargement may not experience decrease to normal size.8
5.4 Treatment goals for hepatomegaly1,9 (Fig. 2)
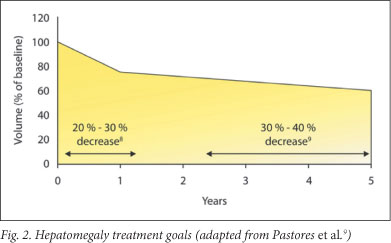
Reduce and maintain the liver volume to/at 1.0 - 1.5 times normal (according to body weight); reduce the liver volume by 20 - 30% within years 1 - 2 and by 30 - 40% by years 3 - 5.
Treatment response may vary. Patients with severe hepatic enlargement may not experience decrease to normal size.9
5.5 Treatment goals for skeletal pathology (Table 3)1'9 Lessen or eliminate bone pain within 1 - 2 years; prevent bone crises; prevent osteonecrosis and subchondral joint collapse; improve bone mineral density (BMD) - adult patients: increase BMD within 3 - 5 years.
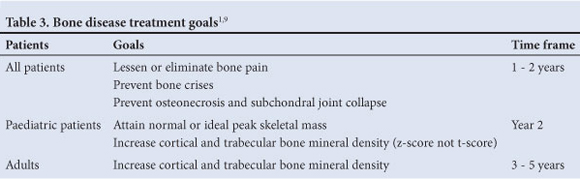
Improve Bone Marrow Burden Score by 50% within 2 - 3 years (measured by MRI).6
5.6 Treatment goals for pulmonary involvement1'9
Reverse hepatopulmonary syndrome and dependency on oxygen; ameliorate pulmonary hypertension (ERT plus adjuvant therapies); improve functional status and quality of life; prevent rapid deterioration of pulmonary disease and sudden death; prevent pulmonary disease by timely initiation of ERT and avoidance of splenectomy.
5.7 Treatment goals for functional health and well-being1'9
Improve or restore physical function for carrying out normal daily activities and fulfilling functional roles
Improve scores from baseline of a validated quality of life instrument within 2 - 3 years or less depending on disease burden.
5.8 Treatment goals for other parameters9
Physical examination results should improve; stabilise or decrease serial measurements of the biomarkers such as chitotriosidase, TRAP, or ACE.
6. Safety information
6.1 Adverse effects of imiglucerase10
Approximately 15% of patients treated and tested have developed IgG antibodies to imiglucerase during the first year of therapy. Patients who developed IgG antibodies usually did so within 6 months of treatment and rarely developed antibodies to imiglucerase after 12 months of therapy. Approximately 46% of patients with detectable IgG antibodies experienced symptoms of hypersensitivity. Patients with antibodies to imiglucerase have a higher risk of hypersensitivity reaction. Conversely, not all patients with symptoms of hypersensitivity have detectable IgG antibody. It is suggested that patients be monitored periodically for IgG antibody formation during the first year of treatment.
Treatment with imiglucerase should be approached with caution in patients who have exhibited symptoms of hypersensitivity to the product. Anaphylactoid reactions have been reported in less than 1% of the patient population. Further treatment with imiglucerase should be conducted with caution in these patients. Most patients have successfully continued therapy after a reduction in the infusion rate and pretreatment with antihistamines and/or corticosteroids.
Adverse event forms should be submitted to the Medicines Control Council and/or the relevant pharmaceutical company.
7. Follow-up assessment (also refer to Section 3.3)
7.1 Timing1
Every 3 months until stable, then 6-monthly:
Clinical evaluation of haematological, visceral, skeletal and, where appropriate, neurological disease and application of a validated health-related quality of life instrument are recommended
Biomarkers including chitotriosidase, ACE and/or TRAP
Blood tests including FBC, liver enzymes and immunoglobulins, as well as other specific investigations depending on baseline results and ongoing co-morbidities.
IgG drug-specific antibody assessment as clinically indicated.
MRI volumetric evaluation of liver and spleen; 2-yearly for stable patients, sooner if indicated.
Imaging of bones as clinically indicated.
7.2 Asplenic patients
Asplenic Gaucher disease patients are at increased risk of osteonecrosis,12,13 pulmonary hypertension,14 cholelithiasis,15 and liver cirrhosis.16 Monitoring therapy by serial measurement of haemoglobin and platelet counts is usually unhelpful. Spleen size clearly cannot be used to monitor response to treatment, therefore particular attention should be paid to systemic and bony symptoms and to biomarkers of disease in order to prevent episodes of bone infarction.
Asplenic patients (especially females) are at increased risk of pulmonary hypertension. Clinicians should maintain a high index of suspicion for this complication. ECG, echocardiogram and Doppler studies should be performed yearly.
Overwhelming sepsis is a principal complication of the asplenic state, mainly as a result of infection with Gram-positive encapsulated organisms (i.e. Streptococcus pneumoniae).17
7.3 Management of asplenic patients1
Ensure that patients who have undergone splenectomy have received the following immunisations:
Haemophilus influenzae type b (Hib)
Pneumococcal polyvalent vaccine repeated every 5 years (for children under 10 years give two doses of pneumococcal conjugate vaccine 2 months apart followed by a dose of polyvalent vaccine 2 months later)
Influenza annually.
Long-term regular antibiotic prophylaxis with penicillin V 250 mg bd or erythromycin 250 mg bd, if allergic to penicillin, is mandatory.
If possible, patients should carry a standard medical information card stating: 'I have no functioning spleen.' Patients should be warned that they are at increased risk of sepsis.
8. Adjuvant therapy1
Adult patients may benefit from bisphosphonate therapy either orally (which can be prescribed by their general practitioner) or intravenously. Little evidence as to the efficacy of this treatment in Gaucher disease is available. Guidelines for the use of bisphosphonates should be similar to those of other causes of osteopenia/osteoporosis.
8.1 Other treatment considerations
Vitamin D, calcium may be considered; specific pain medication;1 seizure/neurological management; pulmonary hypertension management.1 There are no known drug interactions with imiglucerase; therefore co-morbidities are not an indication to withhold therapy.10
9. Surgical management1
Orthopaedic surgical intervention is commonly required to restore function and correct deformity. Subchondral bone collapse as a result of avascular necrosis leads to pain and loss of function and may require joint replacement, most commonly of the hip joint, but also of the knee and shoulder.18 Given that joint replacement is often carried out at a young age, there is frequent requirement for complex revision surgery.
Gallstone disease is also more prevalent in Gaucher disease.13 Peri-operative medical management of Gaucher disease patients must take account of: (i) the risk of infection in the asplenic patient; and (ii) the risk of bleeding associated with thrombocytopenia, platelet function defects and coagulopathies associated with the condition.
10. Indications for cessation of treatment1
Specific treatment may be withdrawn, following careful discussion with the patient and the multidisciplinary team, under the following circumstances:
Intolerable and unavoidable adverse effects; intercurrent illness.
Where long-term quality of life or expected survival is such that the patient will gain no significant benefit from specific treatment for Gaucher disease.
Lack of responsiveness to treatment, having made all appropriate dose adjustments and measures to improve effectiveness of treatment. This applies in the unlikely event of complete resistance to treatment and to irreversible progression of individual aspects of Gaucher disease (most likely neurological) whereby the patient's quality of life is very poor and where there is little or no prospect of response to treatment.
At the request of the patient, or properly allocated guardian acting in the patient's best interests, if the patient is properly deemed not competent.
If the circumstances of the patient's lifestyle are such that sufficient compliance with treatment is not possible. Such cases might include intravenous drug abuse associated with a peripatetic lifestyle.
If the health and well-being of medical and/or nursing staff are placed under significant threat as a result of the actions or lifestyle of the patient.
11. Role of the Lysosomal Storage Disorder Medical Advisory Board
11.1 Interaction between treating physicians and the LSD MAB
The MAB will act as consultants to assist treating physicians in the management of their patients.
11.2 Interaction between medical aid organisations and the LSD MAB
Medical aids/funders may consult the MAB regarding patients new to treatment or when dosage adjustments are requested.
Disclaimer. These guidelines were prepared for physicians and other healthcare professionals on behalf of the LSD MAB and reflect the best data available at the time. Caution should be exercised in interpreting the data; future studies may require alteration of the conclusions or recommendations in this report. It may be necessary or even desirable to depart from the guidelines in the interests of specific patients and special circumstances. These guidelines do not represent all the possible methods of management applicable to all patients, do not exclude any other reasonable methods, and will not ensure successful treatment in every situation. The unique circumstances of each patient must be taken into account by the responsible physician making decisions on any specific therapy. Just as adherence to these guidelines may not constitute defence against a claim of negligence, so deviation from them should not necessarily be deemed negligent.
Acknowledgements. The contributions of the MAB members are gratefully acknowledged: Dr Louisa Bhengu (medical geneticist, University of the Witwatersrand), Prof. Alan Davidson (paediatric haematologist-oncologist, University of Cape Town), Dr Paul du Toit (physician, private practice), Dr Trevor Gerntholtz (nephrologist, private practice), Dr Kenny Govendragaloo (paediatric cardiologist, private practice), Dr Rene Heitner (paediatrician, private practice), Dr Bertram Henderson (medical geneticist, University of the Free State), Dr Lawrence Mubaiwa (paediatric neurologist, University of KwaZulu-Natal), Dr Sheeba Varughese (paediatrician, University of the Witwatersrand).
Guideline sponsor. The meetings of the LSD MAB were sponsored by Genzyme.
Conflict of interest. The MAB was sponsored by Genzyme.
References
1. Deegan P, Hughes D, Mehta A, Cox TM. UK National Guideline for Adult Gaucher Disease, April 2005. Available from http://www.specialisedservicesnhs.uk./library/23/Guidelines_for_AdultGauchers_Disease.pdf (accessed 24 August 2011). [ Links ]
2. Vellodi A, Wraith JE, McHugh K, Cooper A. Guidelines for the management of Paediatric Gaucher Disease in the United Kingdom. Available from http://www.specialisedservices.nhs.uk/library/23/Guidelines_for_Paediatric_Gauchers_Disease.pdf (accessed 24 August 2011). [ Links ]
3. Grabowski GA, Kacena K, Cole JA, et al. Dose-response relationships for enzyme replacement therapy with imiglucerase/alglucerase in patients with Gaucher disease type 1. Genet Med 2009;11(2):92-100. [http://dx.doi.org/10.1097%2FGIM.0b013e31818e2c19] [ Links ]
4. Sims KB, Pastores GM, Weinreb NJ, et al. Improvement of bone disease by imiglucerase (Cerezyme) therapy in patients with skeletal manifestations of type 1 Gaucher disease: results of a 48-month longitudinal cohort study. Clin Genet 2008;73:430-440. [http://dx.doi.org/10.1111%2Fj.1399-0004.2008.00978.x] [ Links ]
5. Mistry PK, Deegan P, Vellodi A, et al. Timing of initiation of enzyme replacement therapy after diagnosis of type 1 Gaucher disease: effect on incidence of avascular necrosis. Br J Haematol 2009;147(4):561-570. [http://dx.doi.org/10.1111%2Fj.1365-2141.2009.07872.x] [PMID:19732054] [ Links ]
6. Maas M, van Kuijk C, Stoker J, Akkerman EM, Aerts JFMG, van Heeten GJ. Quantification of bone involvement in Gaucher disease: MR imaging bone marrow burden score as an alternative to Dixon quantitative chemical shift MR imaging - initial experience. Radiology 2003;229(2):554-561. [http:// dx.doi.org/10.1148%2Fradiol.2292020296] [ Links ]
7. Ida H, Rennert OM, Ito T, et al. Type 1 Gaucher disease: phenotypic expression and natural history in Japanese patients. Blood Cells Mol Dis 1998;24(1):73-81. [http://dx.doi.org/10.1006%2Fbcmd.1998.0172] [ Links ]
8. Beutler E, Demina A, Laubscher K, et al. The clinical course of treated and untreated Gaucher disease: a study of 45 patients. Blood Cells Mol Dis 1995;21(2):86-108. [http://dx.doi.org/10.1006%2Fbcmd.1995.0012] [ Links ]
9. Pastores GM, Weinreb NJ, Aerts H, et al. Therapeutic goals in the treatment of Gaucher disease. Semin Hematol 2004;41(suppl 5):4-14. [http://dx.doi.org/10.1053%2Fj.seminhematol.2004.07.009] [ Links ]
10. Cerezyme approved package insert. Genzyme, February 2010. [ Links ]
11. Granovsky-Grisaru S, Belmatoug N, vom Dahl S, et al. The management of pregnancy in Gaucher disease. Eur J Obstet Gynecol Reprod Biol 2011;156:3-8. [http://dx.doi.org/10.1016%2Fj.ejogrb.2010.12.024] [ Links ]
12. Rodrigue SW, Rosenthal DI, Barton NW et al. Risk factors for osteonecrosis in patients with type 1 Gaucher's disease. Clin Orthop 1999;362:201-207. [http://dx.doi.org/10.1097%2F00003086-199905000-00029] [ Links ]
13. Mankin HJ, Rosenthal DI, Xavier R. Gaucher disease. New approaches to an ancient disease. J Bone Joint Surg Am 2001;83-A(5):748-762. [ Links ]
14. Mistry PK, Sirrs S, Chan A, et al. Pulmonary hypertension in type 1 Gaucher's disease: genetic and epigenetic determinants of phenotype and response to therapy. Mol Genet Metab 2002;77:91-98. [http://dx.doi.org/10.1016%2FS1096-7192%2802%2900122-1] [ Links ]
15. Rosenbaum H, Sidransky E. Cholelithiasis in patients with Gaucher disease. Blood Cells Mol Dis 2002;28(1):21-27. [http://dx.doi.org/10.1006%2Fbcmd.2001.0480] [ Links ]
16. Lachmann RH, Wight DGD, Lomas DJ, et al. Massive hepatic fibrosis in Gaucher's disease: clinic- pathological and radiological features. QJM 2000;93:237-244. [http://dx.doi.org/10.1093%2Fqjmed%2F93.4.237] [ Links ]
17. Ein SH, Shandling B, Simpson JS, et al. The morbidity and mortality of splenectomy in childhood. Ann Surg 1977;185(3):307-310. [http://dx.doi.org/10.1097%2F00000658-197703000-00010] [ Links ]
18. Tauber C, Tauber T. Gaucher disease - the orthopaedic aspect. Report of seven cases. Arch Orthop Trauma Surg 1995;114(3):179-182. [http://dx.doi.org/10.1007%2FBF00443393] [ Links ]
 Correspondence to:
Correspondence to:
A Davidson
(alan.davidson@uct.ac.za)


{kind=link}