










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSAMJ: South African Medical Journal
On-line version ISSN 2078-5135
Print version ISSN 0256-9574
SAMJ, S. Afr. med. j. vol.102 n.8 Pretoria Aug. 2012
RESEARCH
Inherited polyglutamine spinocerebellar ataxias in South Africa
D C SmithI; A BryerII; L M WatsonIII; L J GreenbergIV
IMSc (Med). Division of Human Genetics, Department of Clinical Laboratory Sciences, Institute of Infectious Disease and Molecular Medicine, Faculty of Health Sciences, University of Cape Town
IIMB BCh, FCP (SA), MMed (Neurology), FC Neurology (SA), PhD. Division of Neurology, Department of Medicine, University of Cape Town and Groote Schuur Hospital, Cape Town
IIIMSc (Med). Division of Human Genetics, Department of Clinical Laboratory Sciences, Institute of Infectious Disease and Molecular Medicine, Faculty of Health Sciences, University of Cape Town
IVPhD. Division of Human Genetics, Department of Clinical Laboratory Sciences, Institute of Infectious Disease and Molecular Medicine, Faculty of Health Sciences, University of Cape Town
ABSTRACT
OBJECTIVE: To determine the frequency and distribution of polyglutamine spinocerebellar ataxias (SCAs) from referrals over a 24-year period to the National Health Laboratory Service (NHLS) in South Africa (SA).
METHODS: Paper-based clinical reports in the University of Cape Town laboratory and the NHLS electronic patient record database spanning a 24-year period were mined for information regarding the molecular diagnosis, ethnicity and CAG repeat length for individuals referred for molecular genetic testing for the polyglutamine SCAs.
RESULTS: SCA1 and 7 are the most frequent types of polyglutamine SCA in the SA patient population, followed by SCA2, 3 and 6. SCA1 is the most common type in the coloured, white and Indian populations, whereas the majority of indigenous black African patients are affected with SCA7 and 2. Of individuals tested, 22% were found to be positive for one of the polyglutamine SCAs.
CONCLUSION: Although trends in the frequency and distribution of the polyglutamine SCAs in SA have not changed significantly since our previous study in 2003, they differ remarkably from those reported elsewhere, and reflect the unique genetic and demographic background of SA. The provision of accurate and complete patient information and family history is crucial to the diagnostic process, to enable comprehensive epidemiological studies and assist in developing therapeutic and patient management strategies.
The spinocerebellar ataxias (SCAs), also referred to as autosomal dominant cerebellar ataxias (ADCAs), are inherited diseases characterised by cerebellar and spinal cord degeneration. There are at least 31 known forms of SCA. The SCAs are clinically heterogeneous, with the age of onset, severity and rate of disease progression differing significantly between individuals.1 Onset of clinical symptoms usually occurs between the third and fifth decade, although cases of onset in early childhood and over 60 years of age have been reported, and typically progress to death within 10 - 20 years.2 A subset of the SCAs is caused by the expansion of a CAG repeat motif in the disease-causing gene, resulting in an expanded polyglutamine tract within the corresponding protein. SCA1, 2, 3, 6, 7 and 17 are therefore additionally classified as polyglutamine diseases. Table 1 summarises the clinical features of the more common types of SCA in South Africa (SA).

The prevalence of the SCAs in Europe is estimated to be 1 - 3 per 100 000.1 Although studies have investigated SCA frequency in other countries, only one has focused on the epidemiology of the polyglutamine SCAs in southern Africa. In 2003, Bryer and colleagues examined the spectrum and frequencies of the polyglutamine SCAs in SA through clinical and molecular evaluation of 54 families.3 This study includes the most recent epidemiological figures of the frequency of the polyglutamine SCAs in SA.
Methods
Patients from the SA public or private health sectors who presented with clinical signs of SCA were referred to the molecular diagnostics laboratory of the National Health Laboratory Service (NHLS) in Cape Town, the only centre in SA offering molecular genetic testing for the polyglutamine SCAs (SCA1, 2, 3, 6, 7 and 17) since testing was initiated in 1987.
The QIAamp DNA Blood Mini kit (Qiagen) was used for genomic DNA isolation from fresh or frozen blood from each individual. The CAG repeat region within each causative gene was amplified using a multiplex PCR method,4 followed by capillary electrophoresis on the ABI 3100 Genetic Analyzer (Applied Biosystems). SCA17 repeat sizes were determined using previously described methods.5 The sizes of the repeats were determined by comparison with the GeneScan 500 Rox Size Standard (Applied Biosystems). Each run included positive control samples of known CAG repeat length. Samples with homozygous alleles in the normal repeat range were not re-tested. The NHLS testing laboratory undergoes annual external quality assessments (EQA) by the European Molecular Genetics Quality Network (EMQN).
Paper-based diagnostic reports in the laboratory at the University of Cape Town (UCT) and the NHLS electronic patient record database for 1987 - 2011 were mined for information regarding the molecular diagnosis, ethnicity (reported by the referring clinician) and CAG repeat length for each individual patient referral. Ethical approval was granted by the institutional Human Research Ethics Committee (HREC REF 229/2010, renewed in 2011). Where applicable, written informed consent was obtained for biological material intended for research purposes.
Results
Distribution of SCAs in SA
Of the 1 113 individuals tested for the panel of 5 polyglutamine SCAs over 24 years, 245 (22%) were identified with pathogenic expanded repeats in 1 of the 5 tested SCA loci (Table 2). With a population of 50.5 million in 2011,6 the annual incidence of the polyglutamine SCAs in SA is therefore estimated to be approximately 2.02/10 000 000 per year. The molecular testing laboratory is not involved in patient management and follow-up, and could not comment on the current level of survival of these patients. Similar to 2003 data,3 SCA1 and 7 accounted for most positive cases (48.8% and 26.6%, respectively). Most individuals affected with SCA2, 3 and 6 were referred as single cases, while many positive for SCA1 or 7 were referred as part of affected families (>2 affected individuals). A single individual was diagnosed with SCA17. Molecular diagnostic testing for SCA17 was introduced in 2005, and is therefore excluded from the analysis of the full 24-year period.
Distribution of the polyglutamine SCAs among SA ethnic groups
SA has diverse population groups of different genetic origins; 79.5% are indigenous black Africans, 9% white, 9% coloured, and 2.5% of Indian or Asian ethnicity.6 The demographic information supplied with molecular diagnostic testing referrals to the UCT laboratory rarely contains detailed biographical or clinical information; therefore the ethnicity of 23.6% families is listed as 'unknown' (Table 3). Within this SA cohort, the coloured and white populations were most commonly diagnosed with SCA1, whereas the highest percentages of black African patients were diagnosed with SCA2 or 7. Individuals of Indian descent were exclusively affected with SCA1 or 2.
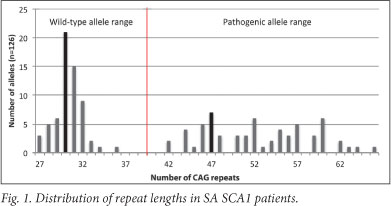
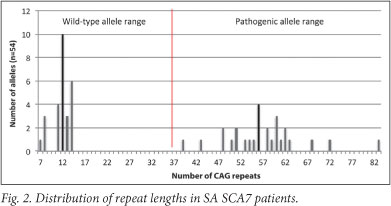
Distribution of repeat lengths
Figs 1 and 2 present the CAG repeat lengths of a subset of SCA1 and 7 patients for whom data were available within the SA SCA patient population. Wild-type SCA1 alleles typically contain between 6 and 38 repeats, where alleles with more than 39 repeats are classified as pathogenic.7 Within the SA SCA1 cohort, the most common wild- type allele contained 30 CAG repeats (range of 27 - 36 repeats), whereas the most common pathogenic allele contained 47 repeats (range of 42 - 66 repeats, Fig. 1). In SCA7, alleles with >36 repeats are classified as pathogenic, while wild-type alleles range in size from 4 to 19 repeats.7 Alleles with 20 - 35 repeats are classified as intermediate alleles. Intermediate alleles are not pathogenic, but may pose a risk to the offspring of the carrier through meiotic expansion into the pathogenic range during vertical transmission.8 Within the SA SCA7 patient group, the most common wild-type allele contained 12 CAG repeats (range of 7 - 14). The most frequent pathogenic allele length was 56, with a pathogenic range of 39 - 83 repeats (Fig. 2).
Discussion
The distribution of the polyglutamine SCAs in SA differs significantly from other countries. The incidence of the polyglutamine SCAs calculated here is likely to be an underestimation, since molecular diagnostic testing referrals from geographically remote areas and low-income communities are limited by absent infrastructure and financial constraints. Of concern is that almost 80% of patients referred for molecular diagnosis tested negative for the polyglutamine SCAs, highlighting that other (possibly undiagnosed) neurological disorders contribute to the health burden in SA. Diagnostic referral forms rarely contain family history information, so it is unclear whether these cases are sporadic or familial. If a significant proportion of them were found to be associated with a positive family history, appropriate measures could be undertaken to identify additional inherited neurological disorders that are common in SA. Future work is likely to include pilot studies to screen patients who have tested negative for the panel of 5 SCAs, for other SCAs. However, SA's unique population composition presents difficulties in predicting the SCAs that are most likely to present in SA, since these may differ significantly from those seen internationally. SCA3 has been reported as the most common of the SCAs, accounting for the majority of ADCA cases in regions of Portugal, the Netherlands, Brazil, China, Japan and Germany.1 By contrast, SCA3 in SA was identified in only 8 individuals representing at least 3 ethnic groups over the 24-year period. SCA1 remains the most frequent polyglutamine SCA in SA,3 where it is overrepresented in the coloured and white population groups.
The frequency of the SCA7 mutation in SA remains one of the highest reported.3 SCA7 is the most common dominant ataxia in Sweden, Norway, Denmark and Finland, as a result of a founder effect.9 A similar experimental approach to that of the Jonasson et al. study9 identified a shared haplotype within the SA SCA7 patient cohort,10 which consists almost entirely of individuals of black African ancestry. Evidence from our research laboratory suggests that this SA SCA7 haplotype may be present in a SCA7 family native to northern Namibia, and a Zambian SCA7 patient, supporting the hypothesis that the founder effect may extend geographically into areas north of SA.
Similarly, 2 distinct haplotypes are associated with the SCA1 mutation in the SA coloured patient population.11,12 The origin and extent of these haplotypes is being investigated; however, they are likely to be present in additional ethnic groups, owing to the heterogeneous composition of the coloured population.
Therapeutically, the shared haplotypes within a specific patient population may be significant, since they increase the likelihood of patients inheriting identical alleles with a single nucleotide polymorphism (SNP) in linkage disequilibrium with the disease-causing mutation. Studies have demonstrated the utility of such disease-linked SNPs as targets for the design of allele-specific RNA interference (RNAi)-based therapies for both SCA112 and 7,13 suggesting that allele-specific therapies may selectively silence mutant gene expression while retaining functional levels of the wild-type copy. Similar approaches are being considered for other neurological disorders such as Huntington's disease.14
Information regarding trends in ethnicities may be valuable in the clinical diagnosis of the polyglutamine SCAs, particularly in cases where molecular diagnostic confirmation is not possible. Individuals of coloured ethnicity appear more likely to be affected with SCA1, possibly due to their two originating founder effects. White individuals are positive for all 5 types of polyglutamine SCA, with most individuals testing positive for SCA1. Individuals of Indian descent in SA have, to date, been solely identified with SCA1 or 2. Reports suggest higher distributions of SCA1 and 2 in various populations native to India, but there are additional reports of Indian individuals diagnosed with SCA3 or 6.15 Until recently, the SCA7 mutation was found exclusively in individuals of black African descent, with the SCA2 mutation arising at a similarly high frequency. The first white SA SCA7 patient was identified early in 2011. Owing to the lack of epidemiological studies on the African continent and within indigenous populations, it is not known whether these trends are representative of other African countries. Information regarding the occurrence of the polyglutamine SCAs in African countries is limited to reports from countries such as Mali.16
The size of the pathogenic CAG repeat in the polyglutamine SCAs is inversely correlated with the age of onset of symptoms and the rate of disease progression.8 Individuals with larger repeat lengths tend to develop symptoms at an earlier age, and progress at a faster rate than patients with smaller repeats. Therefore, knowing the repeat size of an affected SCA individual may aid in their clinical management. Historically, the exact repeat size of SCA patients has not been recorded as part of the diagnostic process, and therefore we could not comment on the distribution of repeat sizes in SA SCA2, 3 and 6 patients. The UCT Division of Human Genetics has ongoing research projects on SCA1 and 7, which allowed for re-testing and sizing of the DNA banked for the SCA1 and 7 patient groups. The repeat size ranges for SA SCA1 and 7 patients lie within those of other population groups,7 although accurate repeat size data were available for only half of each patient cohort, since repeat sizes were not systematically recorded for the full 24-year period. The lack of large normal alleles in these groups (alleles within the normal range, but with a larger repeat length) also supports the hypothesis that the expanded alleles are prevalent because of a founder effect, rather than the independent expansion of large normal alleles. There is a close association between the frequency of large normal alleles and the prevalence of the SCAs within a given population,17 which may further explain the reduced prevalence of the polyglutamine SCAs in SA. However, detailed studies on repeat sizes of unaffected SA individuals are needed to support this hypothesis.
Our study encountered a paucity of information on ethnicity and CAG repeat-length data. This highlights the need for more detailed diagnostic reports and patient information to improve the accuracy of epidemiological studies and to assist in patient management. Detailed family history data should also be obtained where possible, as multi-generation pedigrees greatly enhance the ability of researchers to assign haplotypes - a critical step in determining future eligibility for emerging RNAi-based therapies. Such research is hampered by the limited availability of biological samples from SCA patients and their unaffected family members. We therefore recommend that medical practitioners encourage SCA families to consider participation in molecular research studies.
Our reported frequency of the polyglutamine SCAs in SA reflects the unique population ethnic and geographical distribution. While there has been no significant change in trends of distribution of the polyglutamine SCAs in SA over the past 8 years, this report highlights the lack of patient information that accompanies molecular diagnostic referrals for genetic testing. Critical patient biographical information, such as family history and ethnic background, will aid researchers to identify SA population trends, resulting in streamlined diagnostic strategies, improved clinical utility and validation of tests and, importantly, reduced costs. This information will also be crucial in identifying patients and families who may benefit from future RNAi-based gene silencing therapies.
Acknowledgements. We thank Dr Karen Fieggen and Ms Fiona Baine for critical review of the manuscript and personal communications, and Dr Rene Goliath and Ms Alina Esterhuizen for genotyping data and diagnostic results. We acknowledge funding from the National Research Foundation (NRF) (opinions and conclusions are those of the authors and are not necessarily attributed to the NRF), German Academic Exchange Service (DAAD-NRF), Harry Crossley Foundation (LW), UCT Research Council, Commonwealth Scholarship Commission UK (LW), and Deutscher Akademischer Austausch Dienst (DS).
References
1. Durr A. Autosomal dominant cerebellar ataxias: polyglutamine expansions and beyond. Lancet Neurol 2010;9:885-894. [http://dx.doi.org/10.1016/S1474-4422(10)70183-6] [ Links ]
2. Orr HT, Zoghbi HY. Trinucleotide repeat disorders. Annu Rev Neurosci 2007;30:575-621. [http:// dx.doi.org/10.1146/annurev.neuro.29.051605.113042] [ Links ]
3. Bryer A, Krause A, Bill P, et al. The hereditary adult-onset ataxias in South Africa. J Neurol Sci 2003;216:47-54. [http://dx.doi.org/10.1016/S0022-510X(03)00209-0] [ Links ]
4. Dorschner MO, Barden D, Stephens K. Diagnosis of five spinocerebellar ataxia disorders by multiplex amplification and capillary electrophoresis. J Mol Diagn 2002;4:108-113. [http://dx.doi.org/10.1016/S1525-1578(10)60689-7] [ Links ]
5. Fujigasaki H, Martin JJ, De Deyn PP, et al. CAG repeat expansion in the TATA box binding protein gene causes autosomal dominant cerebellar ataxia. Brain 2001;124:1939-1947. [http://dx.doi.org/10.1093/brain/124.10.1939] [ Links ]
6. Statistics South Africa. Statistical release P0302 mid-year population estimates, 2011. http://www.statssa.gov.za/publications/P0302/P03022011.pdf (accessed 1 December 2011). [ Links ]
7. Schöls L, Bauer P, Schmidt T, et al. Autosomal dominant cerebellar ataxias: clinical features, genetics, and pathogenesis. Lancet Neurol 2004;3:291-304. [http://dx.doi.org/10.1016/S1474-4422(04)00737-9] [ Links ]
8. Sequeiros J, Martindale J, Seneca S. EMQN Best Practice guidelines for molecular genetic testing of SCAs. Eur J Hum Genet 2010;18:1188-1195. [http://dx.doi.org/10.1038/ejhg.2010.8] [ Links ]
9. Jonasson J, Juvonen V, Sistonen P, et al. Evidence for a common Spinocerebellar ataxia type 7 (SCA7) founder mutation in Scandinavia. Eur J Hum Genet 2000;8:918-922. [http://dx.doi.org/10.1038/sj.ejhg.5200557] [ Links ]
10. Greenberg J, Solomon G, Vorster A, et al. Origin of the SCA7 gene mutation in South Africa: implications for molecular diagnostics. Clin Genet 2006;70:415-417. [http://dx.doi.org/10.1111/ j.1399-0004.2006.00680.x] [ Links ]
11. Ramesar RS, Bardien S, Beighton P, et al. Expanded CAG repeats in spinocerebellar ataxia (SCA1) segregate with distinct haplotypes in South African families. Hum Genet 1997;100:131-137. [http://dx.doi.org/10.1007/s004390050478] [ Links ]
12. Baine FK. Identification of a suitable SNP for allele-specific silencing of the disease-causing gene in SCA1 patients in South Africa. MSc dissertation, University of Cape Town, 2010. [ Links ]
13. Scholefield J, Greenberg LJ, Weinberg MS, et al. Design of RNAi hairpins for mutation-specific silencing of ataxin-7 and correction of a SCA7 phenotype. PLoS One 2009;4:e7232. [http://dx.doi.org/10.1371/journal.pone.0007232] [ Links ]
14. Takahashi M, Watanabe S, Murata M, et al. Tailor-made RNAi knockdown against triplet repeat disease-causing alleles. Proc Natl Acad Sci USA 2010;107:21731-21736. [http://dx.doi.org/10.1073/pnas.1012153107] [ Links ]
15. Basu P, Chattopadhyay B, Gangopadhaya PK, et al. Analysis of CAG repeats in SCA1, SCA2, SCA3, SCA6, SCA7 and DRPLA loci in spinocerebellar ataxia patients and distribution of CAG repeats at the SCA1, SCA2 and SCA6 loci in nine ethnic populations of eastern India. Hum Genet 2000;106:597-604. [http://dx.doi.org/10.1007/s004390050031] [ Links ]
16. Traoré M, Coulibaly T, Meilleur KG, et al. Clinical and genetic analysis of spinocerebellar ataxia in Mali. Euro J Neurol 2011;18:1269-1271. [http://dx.doi.org/10.1111/j.1468-1331.2011.03376.x] [ Links ]
17. Takano H, Cancel G, Ikeuchi T, et al. Close associations between prevalences of dominantly inherited spinocerebellar ataxias with CAG-repeat expansions and frequencies of large normal CAG alleles in Japanese and Caucasian populations. Am J Hum Genet 1998;63:1060-1066. [http://dx.doi.org/10.1086/302067] [ Links ]
Accepted 11 April 2012.
 Correspondence to:
Correspondence to:
L Greenberg
(jacquie.greenberg@uct.ac.za)

