










Servicios Personalizados
Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkSAMJ: South African Medical Journal
versión On-line ISSN 2078-5135
versión impresa ISSN 0256-9574
SAMJ, S. Afr. med. j. vol.102 no.8 Pretoria ago. 2012
FORUM
HISTORY OF MEDICINE
Loeys-Dietz syndrome: a possible solution for Akhenaten's and his family's mystery syndrome
Ahad Eshraghian; Bart Loeys
Department of Internal Medicine, Shiraz University of Medical Science, Shiraz, Iran. Bart Loeys is from the Center for Medical Genetics, Antwerp University Hospital, Antwerp, Belgium
ABSTRACT
The presence of a familial disease among royal members of 18th dynasty of the new kingdom who ruled in Egypt from the mid-16th to the early 11th centuries BC has been established, largely prompted by the bizarre body shape of Akhenaten (the iconoclastic pharaoh of this dynasty) and his family, as demonstrated in statues and artwork. It had been thought previously that this was an expression of a revolutionised artistic style that followed radical reforms by Akhenaten of Egyptian society, but recent studies on mummies confirmed the presence of a constellation of corresponding pathologies. Several illnesses have been suggested to solve this enigma; we propose Loeys-Dietz syndrome as a probable diagnosis for this genetic affliction within the royal family.
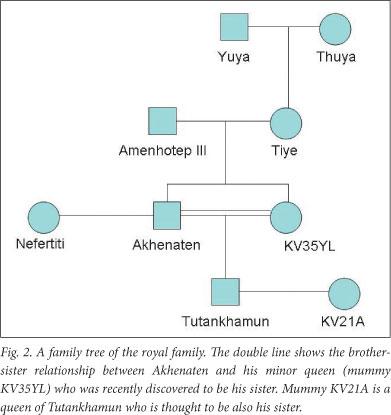
The history of ancient Egypt holds many unsolved, fascinating mysteries. The 18th dynasty is the most famous and glorious of ancient Egyptian dynasties that ruled in Egypt form the mid-16th to the early 11th centuries BC. Many well-known pharaohs such as Hatshepsut, Thutmose III and Tutankhamun belong to this dynasty. Ancient Egypt had large territories and experienced a magnificent period of economic and social development. One little-known aspect of this dynasty that has been the subject of debate is the presence of a familial disease, a notion originating from several statues, sculptures and reliefs of Akhenaten and his family that show elongated heads, faces and extremities, and an underdeveloped thorax with gynaecomastia (Fig. 1). Tutankhamun's death at a young age tends to support this theory, as this might have been indicative of a genetic disorder, especially because royal family marriages between brothers and sisters were frequent, to preserve the royal bloodline, which increased the risk of inherited disorders (Fig. 2).

Several hypotheses have been proposed to explain a familial disease that caused the bizarre appearance of the royalty, including Marfan syndrome, Wilson-Turner X-linked mental retardation syndrome, Fröhlich syndrome (adiposogenital dystrophy), Klinefelter syndrome, androgen insensitivity syndrome,1 myotonic dystrophy,2 and aromatase excess syndrome in conjunction with sagittal craniosynostosis syndrome, or Antley-Bixler syndrome.3 Hawass and co-workers published results of their molecular genetic studies on mummified remains of the royal family of the 18th dynasty to determine any pathologies and familial disease that might have afflicted them.4 Tests showed no sign of craniosynostoses and Antley-Bixler or Marfan syndrome, and could find no evidence of gynaecomastia because the anterior chest wall of the mummies was not available. Therefore, the presence or absence of gynaecomastia is not clear. The feminised appearance of Akhenaten was excluded by the pelvic bone shape. The study group also found evidence of Plasmodium falciparum infection in the mummies. They concluded that none of the presumed hypotheses about the familial disease was correct, but found a constellation of deformities in Tutankhamun and his family. They did not propose any specific diagnoses or syndromes that could explain repeated deformities and pathologies in the members of royal family.
We hypothesise that the 18th dynasty royal family of the new Egyptian kingdom might have suffered from Loeys-Dietz syndrome, which might solve the medical mystery of the 18th dynasty. We emphasise that previous theories regarding the royal family familial illness mainly originated from reliefs, sculptures and limited medical facts obtained from limited studies. Our postulation is based on pathological findings in a recent study on mummies.4
Discussion
Loeys-Dietz syndrome (LDS) is an autosomal dominant inherited disorder of connective tissue that has been recently introduced as an important cause of familial aortic aneurysm. LDS is caused by heterozygous mutations in genes encoding transforming growth factor â receptors 1 and 2 (TGFBR1 and TGFBR2).5 Patients affected with this syndrome are characterised by specific clinical features involving the vascular and skeletal systems as well as craniofacial morphology and skin appearance.
LDS type 1 accounts for nearly 75% of patients with specific craniofacial dysmorpology. In patients with LDS type 2, skin abnormalities are prominent features, and they do not have major craniofacial involvement. Diagnosis is confirmed by detection of mutations in TGFBR1 and TGFBR2.6
LDS is characterised by 4 major categories of clinical features:
1. Cardiovascular system. The most important finding in LDS patients is dilatation of the aorta at the level of the Valsalva sinuses, while aneurysm of the ascending or descending aorta is seen less frequently.6 Congenital heart diseases such as atrial septal defect (ASD), bicuspid aortic valve and patent ductus arteriosus (PDA) are also more prevalent in LDS patients.7 Mitral valve prolapse and regurgitation along with cardiomyopathy secondary to coronary artery dysplasia have been reported in these patients.8
2. Skeletal findings. Joint hyperlaxity, arachnodactyly, pecus excavatum or carinatum, scoliosis and contractures of the feet (talipes equinovarus) are seen in LDS patients, and are also common in Marfan syndrome. Pes planus, spine instability and spondylolisthesis are recurrent findings in this syndrome.6 Dolichostenomelia (long limbs, leading to an increase in the arm span-to-height ratio and a decrease in the upper-to-lower segment ratio) is less common in this syndrome.9
3. Facial dysmorphology. Abnormal facial features are prominent in type II LDS patients. Hypertelorism (wide-set eyes) and a cleft palate or a bifid uvula are characteristic of LDS. Craniosynostosis in the forms of dolichocephaly, bracycephaly and trigonocephaly is common.6 Other craniofacial features are retrognathia, blue sclerae, highly arched palate and dental crowding.10 In type II LDS patients, facial findings are not so severe and include a tall, broad forehead; frontal bossing; a high anterior hair line; hypoplastic supraorbital margins; and a myopathic face.11
4. Skin findings. These patients have velvety and translucent skin with easy bruising and delayed wound healing. Skin manifestations without striking facial abnormalities are seen in type II LDS.6
Akhenaten: The apostate
Amenhotep IV, the strangest ruler of ancient Egypt, was the son of Amenhotep III and his queen Tiye. Some Egyptologists have considered him a criminally mad ruler, while others have admired him as a great reformist. His radical and extensive reform of Egyptian society influenced religion, art, politics and other aspects of Egyptian life. He is probably the first and the only pharaoh who practised monotheism and tried to propagate this doctrine in and around the Egyptian territories. He changed his name to Akhenaten (he who serves Aten) and began to replace images of Amon (the great Egyptian god) and other statues of gods and goddesses with his single god named Aten, whose image was a sun disk. When conflict increased, he left Thebes (the ancient capital of Egypt) and moved to a new region in Middle Egypt, near Amarna village in modern Egypt, and built a new capital named Akhet-Aten (Horizons of the sun disk). He forbade depicting any other god or goddess, and destroyed or seized their temples, which led to a religious war within the country.
Amarna artistic style
Sculptors in ancient Egypt showed kings and royalty in an idealised fashion, and did not show physical defects. But in the new art style that was a consequence of Akhenaten's reforms, artists were free to depict reality, such as an unattractive body, and even exaggerate these features (as cartoonists tend to do).12 Therefore, it is possible that Akhenaten really had an elongated face, underdeveloped thorax with gynaecomastia and a prominent abdomen, and current statues and sculptures are exaggerated forms of this unusual appearance.
Is Akhenaten's religion a key to his disease?
Aten was the name of Akhenaten's god that was reflected as a sun disk. The sun was the symbol of Aten, whose temples had no roof so that sunshine could play directly onto the altar. Akhenaten spent all of his reign propagating Aten throughout Egypt. In a society with traditional and religious prejudices and a long history of numerous deities, what made him introduce one supreme god and eliminate all others? Some have argued that Aten was a political strategy to confine the power of Amon (King of the gods) and his priests. However, as reflected in papyruses, Akhenaten truly adored his god and spent much of his daily routine worshipping Aten, naked, in sunlight, to receive his bliss (Fig. 3). One might also think that Akhenaten had an illness that was alleviated by exposure to sunshine.

Can Loeys-Dietz syndrome justify the pathologies in Akhenaten and his family?
Different syndromes and diseases have been hypothesised to explain a probable underlying disease in Akhenaten, Tutankhamun and 18th dynasty royalty. Marfan syndrome was suspected owing to the marfanoid features of Akhenaten and his family in statues. Recently, however, Marfan syndrome and similar diseases were excluded as none of the royalty fulfilled Marfan criteria.4 However, the latter study confirmed the presence of a group of pathologies in royal members of the 18th dynasty.4 LDS is a clinical continuum and should be suspected in patients with Marfan-like features not fulfilling Ghent criteria.13
Hawass et al. found repeatedly in the royal family, not only clinical features of LDS, but also scoliosis, club foot, incisional hernia, cleft palate, retrognathism, dental crowding and highly arched palate.4 No mummy showed dolichostenomalia, whilst in LDS, in contrast to Marfan syndrome, dolichostenomalia is not frequent. According to their findings, Akhenaten and Tutankhamun had brachycephaly, which can occur in LDS patients (Table 1).
Cardiac pathologies in LDS, such as ASD and cardiomyopathy, are prevalent, and both can induce pulmonary hypertension and right-sided heart failure.14 Patients with right-sided heart failure will develop liver congestion that can progress to cirrhosis over time. Patients with right-sided heart failure even without cirrhosis may have ascites, gynaecomastia and peripheral oedema. Therefore the unusual shape of Akhenaten in statues and paintings of a prominent abdomen and large breasts could be due to ascites and gynaecomastia, which are seen in cirrhosis secondary to right-sided heart failure in the context of LDS; this can be a reason for gynaecomastia if proved to be present.
Vitamin D deficiency has been described in patients with LDS.15 Diffuse musculoskeletal pain and even myopathy, chronic fatigue and depression can occur in patients with hypovitaminosis D.16 Exposure to sunlight is the main source of vitamin D, and treatment of vitamin D deficiency with sun exposure has been reported.17
TGFBR gene polymorphism is associated with bone mineral density and bone turnover, and osteopenia/osteoporosis may also occur in LDS.18 Furthermore, patients with LDS suffer from some skin manifestations as described above. TGF-â signalling pathway in the cells is initiated by binding of the ligand to the TGFBR 2 and subsequent phosphorylation of TGFBR 1. Activated TGFBR 1/TGFBR 2 complex mediates transcription of multiple genes via its downstream Smad signalling proteins. Mutations in TGFBR 1/ TGFBR 2 as seen in LDS enhance the TGF-â signalling pathway and increase transcription of multiple downstream genes and proteins.5 Quan et al. showed that solar ultraviolet irradiation reduces collagen in human skin which is mediated by blocking TGFBRII/Smad signalling.19 This finding was also confirmed in other studies.
The foregoing findings may provide reasons to justify the association between Akhenaten's disease and his religious reforms. Akhenaten might have suffered from chronic musculoskeletal pain owing to vitamin D deficiency and osteoporosis secondary to LDS, and sunshine exposure might have relieved his pain. Furthermore, the diseased TGF-â signalling pathway was down-regulated by his sunshine worshipping of Aten. In this way, he indeed received physical and spiritual beneficence from his god, which strengthened his faith in Aten despite considerable political, religious and social pressures.
Both types of LDS are generally believed to be two sides of a clinical spectrum. Therefore, an affected individual could have no striking facial appearance. Developmental delay has been reported to occur in a small minority of patients with LDS, but most have normal intellectual ability. Only 25% of these patients have an affected parent, and 75% of patients developed de novo mutations. So it would not be unusual if Akhenaten, with the more severe form of the syndrome, had apparently normal parents or children. Another possibility is that Tutankhamun was more severely affected than his father Akhenaten, which may be explained by homozygocity for the TGFBR mutation, because of the consanguinity.
The main cause of early mortality (mean age 26.1 years) in LDS is dissection of a dilated aorta. Although confirmation of aortic aneurysm in the remaining mummies is not possible, neither Akhenaten and Tutankhamun nor other relatives were more than 50 years old, as determined by DNA studies.
Lastly: a related syndrome with aortic aneurysm and early-onset osteoarthritis was recently found to be caused by mutations in the SMAD3 gene. This gene encodes some proteins that are involved in the TGFB pathway. Patients with this condition also presented with bifid uvula/cleft palate, hypertelorism and skin abnormalities.20
Conclusion
Based on the foregoing findings, the possibility of Loeys-Dietz syndrome or an abnormality in the SMAD3 gene should be considered as the underlying disease of this Egyptian royal family. Mutations in TGFBR1 and TGFBR2 or SMAD3 from DNA of remaining mummies can clarify one of the mysteries of Egyptian history.
References
1. Walshe JM. Tutankhamun: Klinefelter's or Wilsons? Lancet 1973;1(7794):109-110. [ Links ]
2. Cattaino G, Vicario L. Myotonic dystrophy in Ancient Egypt. Eur Neurol 1999;41(2):59-63. [ Links ]
3. Braverman IM, Redford DB, Mackowiak PA. Akhenaten and the strange physiques of Egypt's 18th dynasty. Ann Intern Med 2009;150(8):556-560. [ Links ]
4. Hawass Z, Gad YZ, Ismail S, et al. Ancestry and pathology in King Tutankhamuns family. JAMA 2010;303(7):638-647. [ Links ]
5. Van Hemelrijk C, Renard M, Loeys B. The Loeys-Dietz syndrome: an update for the clinician. Curr Opin Cardiol 2010; 25(6):546-551. [ Links ]
6. Loeys BL, Schwarze U, Holm T, et al. Aneurysm syndromes caused by mutations in the TGF-beta receptor. N Engl J Med 2006;355(8):788-798. [ Links ]
7. Attias D, Stheneur C, Roy C, et al. Comparison of clinical presentations and outcomes between patients with TGFBR2 and FBN1 mutations in Marfan syndrome and related disorders. Circulation 2009;120:2541-2549. [ Links ]
8. Eckman PM, Hsich E, Rodriguez ER, Gonzalez-Stawinski GV, Moran R, Taylor DO. Impaired systolic function in Loeys-Dietz syndrome: a novel cardiomyopathy? Circ Heart Fail 2009;2:707-708. [ Links ]
9. Jamsheer A, Henggeler C, Wierzba J, et al. A new sporadic case of early-onset Loeys-Dietz syndrome due to the recurrent mutation p.R528C in the TGFBR2 gene substantiates interindividual clinical variability. J Appl Genet 2009;50:405-410. [ Links ]
10. Yetman AT, Beroukhim RS, Ivy DD, Manchester D. Importance of the clinical recognition of Loeys-Dietz syndrome in the neonatal period. Pediatrics 2007;119:e1199-e1202. [ Links ]
11. Ades LC. Evolution of the face in Loeys-Dietz syndrome type II: longitudinal observations from infancy in seven cases. Clin Dysmorphol 2008;17:243-248. [ Links ]
12. Huppertz A, Wildung D, Kemp BJ, et al Nondestructive insights into composition of the sculpture of Egyptian Queen Nefertiti with CT. Radiology 2009;251(1):233-240. [ Links ]
13. Mizuguchi T, Collod-Beroud G, Akiyama T, et al. Heterozygous TGFBR2 mutations in Marfan syndrome. Nat Genet 2004;36:855-860. [ Links ]
14. Vogel M, Berger F, Kramer A, Alexi-Meshkishvili V, Lange PE. Incidence of secondary pulmonary hypertension in adults with atrial septal or sinus venosus defects. Heart 1999;82:30. [ Links ]
15. Kirmani S, Tebben PJ, Lteif AN, et al. Germline TGF-beta receptor mutations and skeletal fragility: a report on two patients with Loeys-Dietz syndrome. Am J Med Genet A 2010;152A(4):1016-1019. [ Links ]
16. Fabbriciani G, Pirro M, Leli C, et al Diffuse muscoskeletal pain and proximal myopathy: do not forget hypovitaminosis D. J Clin Rheumatol 2010;16(1):34-37. [ Links ]
17. Chandra P, Agarwal M, Sharma SG, Basra S. Tanning can be an alternative source of vitamin d in high risk populations. J Nutr Sci Vitaminol (Tokyo) 2008;54(1):105. [ Links ]
18. Lau HH, Ho AY, Luk KD, Kung AW. Transforming growth factor-beta1 gene polymorphisms and bone turnover, bone mineral density and fracture risk in southern Chinese women. Calcif Tissue Int 2004;74(6):516-521. [ Links ]
19. Quan T, He T, Kang S, Voorhees JJ, Fisher GJ. Solar ultraviolet irradiation reduces collagen in photoaged human skin by blocking transforming growth factor-beta type II receptor/Smad signaling. Am J Pathol 2004;165(3):741-751. [ Links ]
20. Van de Laar IM, Oldenburg RA, Pals G, et al. Mutations in SMAD3 cause a syndromic form of aortic aneurysms and dissections with early-onset osteoarthritis. Nat Gene 2011:43,121-126. [ Links ]
 Correspondence to:
Correspondence to:
A Eshraghian
(eshraghiana@yahoo.com)


{kind=link}