










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSAMJ: South African Medical Journal
On-line version ISSN 2078-5135
Print version ISSN 0256-9574
SAMJ, S. Afr. med. j. vol.102 n.7 Pretoria Jul. 2012
RESEARCH
Confirmation of the recurrent ACVR1 617G>A mutation in South Africans with fibrodysplasia ossificans progressiva
Collet DandaraI; Chris ScottII; Mike UrbanIII; Karen FieggenIV; Regan ArendseV; Peter BeightonVI
IPhD. Division of Human Genetics, Faculty of Health Sciences, University of Cape Town Medical School
IIMB ChB, FCPaed, Cert Medical Genetics. Division of Human Genetics, Faculty of Health Sciences, University of Cape Town Medical School
IIIOMB, MD, PhD, FRCP, FRSSA. Division of Human Genetics, Faculty of Health Sciences, University of Cape Town Medical School
IVFCPaed (SA). Department of Paediatrics, Red Cross War Memorial Children's Hospital and University of Cape Town Medical School
VFCPSA, Cert Medical Genetics. Division of Molecular Biology and Human Genetics, Faculty of Health Sciences, Stellenbosch University
VIPhD, FCPSA. Division of Rheumatology, University of Cape Town and Groote Schuur Hospital
ABSTRACT
OBJECTIVE. Fibrodysplasia ossificans progressiva (FOP) is a rare genetic condition in which progressive ossification of fibrous tissue, tendons and ligaments leads to severe physical handicap. Most affected individuals who have been studied have a recurrent 617G>A mutation in the ACVR1/ALK2 gene that codes for activin A type 1 receptor/activin-like kinase 2. The majority of publications on the genetics of FOP have concerned whites or Asians, and no genetic information is available concerning sub-Saharan blacks. The aim of the project was to determine whether or not this mutation is present in affected persons in South Africa.
METHOD. Molecular mutational analysis was undertaken on genomic DNA from peripheral blood leukocytes from 6 affected South Africans of different population groups (4 Xhosa, 1 coloured, 1 white).
RESULTS. The 6 persons with FOP were all heterozygous for the ACVR1/ALK2 617G>A mutation. This mutation was absent in 6 controls.
CONCLUSION. Confirmation of the presence of this recurrent mutation facilitates diagnostic accuracy in affected persons in South Africa, and allows researchers to narrow the search for molecular targets for rational intervention to the ACVR1/ALK2 domain.
Fibrodysplasia ossificans progressiva (FOP) [OMIM 135100] is a rare genetic disorder in which ossification of connective tissue leads to severe disability. It is an autosomal dominant trait and affected persons have mutations in the activin A type 1 receptor gene (ACVR1), chromosomal locus 2q23-24.1 ACVR1 is one of the f4 type 1 receptors that mediate in the highly conserved bone morphogenetic protein (BMP) signalling pathway, through a domain rich in glycine and serine (GS-domain) residues.2,3
Diagnosing FOP has depended on recognising characteristic clinical and radiological features, and until recently the causative mechanism remained elusive.2,3 FOP molecular investigations has revealed that most affected individuals have the same single nucleotide change 617G>A in the ACVR1 gene. The base change is a missense mutation that leads to the substitution of arginine with histidine (R206H). The point mutation 617G>A in ACVR1 that occurs in the GS-domain alters the ligand-dependent sensitivity for BMP signalling in connective tissue progenitor cells. Under normal circumstances, type 1 receptors such as ACVR1 are inactive until stimulated by extracellular BMPs through phosphorylation.1-3
FOP diagnosis is often delayed owing to its rarity. The clinical manifestations may also be confused with those of arthrogryposis multiplex congenita, several genetic rigid-joint syndromes, and various forms of childhood rheumatic disease.
As most genetic analyses have been carried out in white and Asian populations, the question arises as to whether or not this specific mutation (617G>A) is associated with FOP in all populations. No sub-Saharan black molecular studies interrogating the genetics of ACVR1/ALK2 and its association with FOP have been reported. Therefore, we investigated whether the commonly reported ACVR1/ ALK2 617G>A recurrent mutation also causes FOP in indigenous South Africans.
Methods
Six persons with FOP (4 Xhosa, 1 coloured and 1 white) were available for molecular investigation (Table 1). Three of them were the subject of reports, including full clinical and radiological descriptions, concerning the management and dental implications of FOP in South Africa.4,5
The main features of FOP in the affected South Africans were consistent with the literature. The severity and rate of progression are variable. FOP often presents at birth with shortening and deviation of the great toes. General health remains good and early development is normal. In mid-childhood, tender subcutaneous lumps may appear, frequently on the upper region of the back, that become ossified, and widespread ectopic ossification develops in the connective tissues. The limbs, neck and jaw become tethered by bands of ossification resulting in immobility. Finally, movements are limited to the external muscle of the eye and the diaphragm. Death usually occurs by middle age from respiratory insufficiency.
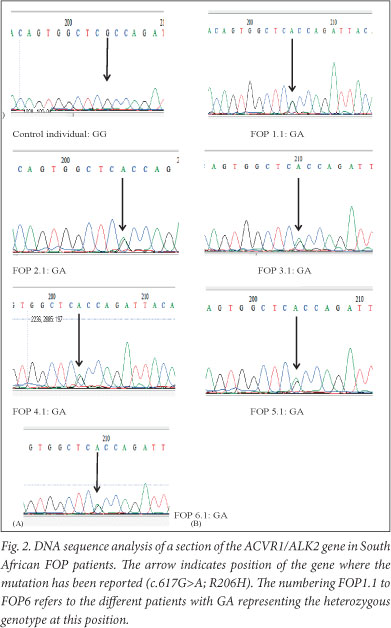
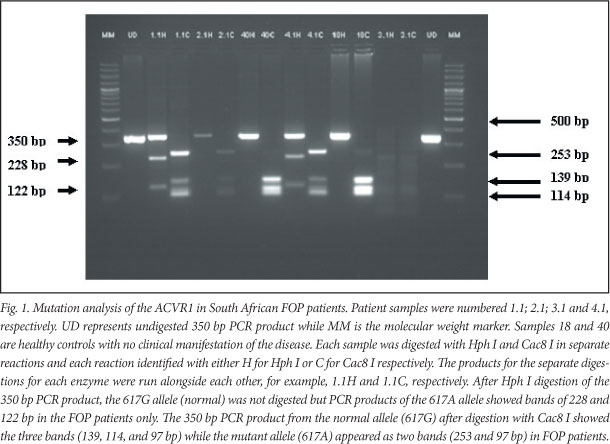
Blood was obtained for molecular genetic analysis from the 6 FOP patients and 6 ethnic-matched controls after obtaining informed consent in accordance with the requirements of the Human Research Ethics Committee, University of Cape Town (ref HREC 026/2010 - PB). Genomic DNA was extracted from peripheral blood leukocytes using a Qiagen DNA extraction kit (Valencia, USA). The single nucleotide polymorphism 617G>A in ACVR1 was analysed according to the method of Shore et al.1 using PCR and restriction fragment length polymorphism (RFLP) using HphI and Cac8l. The 617G>A mutation in ACVR1 eliminates a Cac8I site and forms a new HphI site (Fig. 1). In addition, each of the samples was subjected to sequence analysis.
Results
The 350 bp PCR product from the normal allele (617G) after digestion with Cac8I showed the 3 bands (139, 114, and 97 bp) while the mutant allele (617A) appeared as 2 bands (253 and 97 bp) in persons with FOP. For HphI, PCR products of the 617G allele (normal) were not digested but PCR products of the 617A allele showed bands of 228 and 122 bp in the FOP patients. All 6 persons with FOP were heterozygous for the 617G>A mutation. The mutation was absent in all 6 controls. Sequence analysis (Fig. 2) revealed the heterozygous genotype in all affected individuals while the controls had the normal homozygous 617G/G genotype.
Discussion
Most individuals with FOP are sporadic, representing new mutations for the determinant gene. Although FOP has a worldwide distribution, there are few previous reports of affected persons in South Africa.6 Four of the 6 persons investigated in Cape Town are Xhosa. Reports from several other parts of the world have confirmed the presence of the 617G>A specific mutation in persons with FOP. However, the concept of 1 specific mutation in ACVR1 is no longer tenable as evidenced by the reports associating 605G>T, 983G>A, 774G>C and 1067G>A with FOP in the absence of the recurrent 617G>A.2,7 Nevertheless, the important feature of these mutations is that their resulting amino acid changes mostly occur in the GS or kinase coding domains.7 This observation makes molecular sense in that different mutations in the ACVR1/ALK2 receptor gene could lead to different clinical manifestations of FOP. By extrapolation, mutations within the ACVR1/ALK2 receptor gene that cause variable ACVR1/ ALK2 receptor activity could also lead to different phenotypes, depending on the sensitivities of the domains in which they occur.2,7
This concept could also argue for the involvement of mutations in ACVR1 receptor gene in other related disorders such as some forms of myositis.2,7
The identification of the recurrent 617G>A mutation in almost all FOP patients worldwide, together with the narrowing of all reported mutations to the GS and kinase domains of the ACVR1/ ALK2 gene, provides a specific target of intervention in a critical signalling pathway. The prime target in FOP would be inhibition of the BMP signalling pathway, using RNA technology or monoclonal antibodies.1,8 In South Africa, confirming the recurrent 617G>A mutation in all 6 patients suggests that genetic analysis can aid the diagnosis of suspected FOP, thereby negating the need for invasive procedures that can accelerate its progression.
Conclusions
Demonstrating that 6 affected South Africans with disparate antecedents have the same worldwide documented 617G>A mutation provides a firm starting point for establishing a diagnostic molecular testing facility for FOP in sub-Saharan Africa. In turn, awareness of FOP and the feasibility of molecular diagnostic confirmation would positively influence its medical management in Africa. The ubiquity of the 617G>A mutation could also facilitate research on identifying molecular intervention strategies that may be applied in FOP patients to slow the progression of the disorder, particularly by targeting the BMP signalling pathway.
Acknowledgements. Collet Dandara and Peter Beighton are grateful to the National Research Foundation (NRF) and the Medical Research Council (MRC) of South Africa for support. Collet Dandara thanks the Research Council of the University of Cape for research support.
References
1. Shore EM, Xu M, Feldman GJ, et al. A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva. Nature Genetics 2006;38:525-527. [ Links ]
2. Kaplan FS, Xu M, Seemann P, et al. Classic and atypical fibrodysplasia ossificans progressiva (FOP) phenotypes are caused by mutations in the bone morphogenetic protein (BMP) type I receptor ACVR1. Hum Mutat 2009a;30:379-390. [ Links ]
3. Kaplan FS, Pignolo RJ, Shore EM. The FOP metamorphogene encodes a novel type I receptor that dysregulates BMP signalling. Cytokine & Growth Factor Reviews 2009b;20:399-407. [ Links ]
4. Scott C, Urban M, Arendse R, Dandara C, Beighton P. Fibrodysplasia ossificans progressiva in South Africa: difficulties in management in a developing country. J Clin Rheumatol 2011;17:38-41. [ Links ]
5. Roberts T, Stephen LXG, Scott C, Urban M, Sudi S, Beighton P. Fibrodysplasia ossificans progressive (FOP) in South Africa: dental implications in 5 cases. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 2011;112:11-18. [ Links ]
6. Connor JM, Beighton P. Fibrodysplasia ossificans progressiva in South Africa: case reports. S Afr Med J 1982;61:404-406. [ Links ]
7. Gregson CL, Hollingworth P, Williams M, et al. A novel ACVR1 mutation in the glycine/serine-rich domain found in the most benign case of a fibrodysplasia ossificans progressiva variant reported to date. Bone 2011;48:654-658. [ Links ]
8. Yu PB, Deng DY, Lai CS, et al. BMP type I receptor inhibition reduces heterotopic ossification. Nature Medicine 2008;14:1363-1369. [ Links ]
Accepted 16 January 2012.
Corresponding author: C Dandara (collet.dandara@uct.ac.za)


{kind=link}
{kind=link}