










Serviços Personalizados
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkSAMJ: South African Medical Journal
versão On-line ISSN 2078-5135
versão impressa ISSN 0256-9574
SAMJ, S. Afr. med. j. vol.102 no.7 Pretoria Jul. 2012
RESEARCH
GST polymorphisms and early-onset coronary artery disease in young South African Indians
Alisa PhulukdareeI; Sajidah KhanII; Devapregasan MoodleyIII; Anil A ChuturgoonIV
IMMedSci. Medical Biochemistry, Nelson R Mandela School of Medicine, University of KwaZulu-Natal, Durban
IIPhD. Medical Biochemistry, Nelson R Mandela School of Medicine, University of KwaZulu-Natal, Durban
IIIPhD. Medical Biochemistry, Nelson R Mandela School of Medicine, University of KwaZulu-Natal, Durban
IVMB ChB, FCP. Department of Cardiology, Nelson R Mandela School of Medicine, University of KwaZulu-Natal, Durban
ABSTRACT
BACKGROUND: Glutathione S-transferases (GSTs) detoxify environmental agents which influence the onset and progression of disease. Dysfunctional detoxification enzymes are responsible for prolonged exposure to reactive molecules and can contribute to endothelial damage, an underlying factor in coronary artery disease (CAD).
OBJECTIVES: We aimed to assess 2 common polymorphic variant isoforms in GSTM1 and GSTP1 of GST in young CAD patients.
METHODS: All patients (N=102) were South Africans of Indian ancestry, a population associated with high CAD risk. A corresponding age-, sex- and race-matched control group (N=100) was also recruited. Frequency of the GSTM1 +/0 (v. +/0 and 0/0) and GSTP1 A105/G105 (v. wild-type A105/A105) genotypes was assessed by differential polymerase chain reaction (PCR) and PCR restriction fragment length polymorphism (PCR-RFLP), respectively.
RESULTS: The GSTM1 0/0 and GSTP1 A105/A105 genotypes occurred at higher frequencies in CAD patients compared with the control group (36% v. 18% and 65% v. 48%, respectively). A significant association with CAD was observed in GSTM1 0/0 (odds ratio (OR)=2.593; 95% confidence interval (CI) 1.353 - 4.971; p=0.0043) and GSTP1 A105/A105 OR=0.6011; 95% CI 0.3803 - 0.9503; p=0.0377). We found a significant association between smoking and CAD; the presence of either of the respective genotypes together with smoking increased the CAD risk (GSTP1 A105 relative risk (RR)=1.382; 95% CI 0.958 - 1.994; p=0.0987 and GSTM1 null RR=1.725; 95% CI 1.044 - 2.851; p=0.0221).
CONCLUSION. Our findings support the association of genotypes GSTM1 0/0 and GSTP1 A /A and smoking with CAD.
Atherosclerosis and thrombosis are the 2 major contributing mechanisms in coronary artery disease (CAD), a major health problem and leading cause of death worldwide. The formation of atherosclerotic lesions is augmented by elevated levels of reactive oxygen species (ROS).1
Endothelial cell injury is initiated by increased shear stress, caused by the combination of high blood viscosity, hypertension and vasoconstriction which may be induced by smoking. The toxic chemicals present in tobacco smoke affect the endothelium and enhance smooth-muscle cell proliferation by inducing platelet adherence to damaged cells.2 Nicotine is a strong activator of the sympathetic nervous system, which releases catecholamines that interfere with haemodynamics, resulting in increased myocardial oxygen demand.3 Cigarette smoke contains a complex mixture of approximately 4 000 chemical species and plentiful oxygen-, nitrogen- and carbon-centred free radicals which increase ROS generation and promote oxidative damage.4
Cells produce ROS through normal metabolic processes and disruption of the mitochondrial electron transport chain.5 Cell-intrinsic mechanisms, such as the glutathione (GSH) antioxidant defence system, act to combat ROS. Disturbances in the endogenous antioxidant defence mechanism or ROS overproduction, however, lead to accumulation and availability of free radicals which induce oxidative damage to biomolecules by lipid peroxidation, protein nitration and DNA adduct formation.
Cellular detoxification systems protect against endo- and exogenous harmful substances. Of particular interest is the super-family of glutathione S-transferases (GSTs), which modulate prostaglandin signalling pathways and oxidative stress.6 GSTs function to detoxify electrophilic toxicants, including those found in cigarette smoke, by facilitating conjugation to GSH.7 However, GST function is influenced by genotypic differences from single nucleotide polymorphisms (SNPs).8
GST genes contain several polymorphic variants occurring at high frequency. In the GSTP1 A105/G105 variant, a single nucleotide change from adenine (A) to guanine (G) at codon 105 results in an isoleucine (Ile) to valine (Val) amino acid change. In the GSTM1 null (0/0) polymorphism, complete deletion of the gene encoding this isoform results in abolished enzymatic activity.9
Several studies assessing genetic susceptibility in response to air pollution and cardiovascular outcomes show that GSTP1 and GSTM1 play a role in ROS modulation.10 Others found an association of GSTM1 0/0 with heart failure.11,12 Black carbon and particulate matter exposure was shown to be associated with increased intracellular and vascular cell adhesion molecules, which promoted atherosclerosis; this association was modified by the presence of the GSTM1 polymorphism.13 GSTP1 and GSTM1 polymorphisms have been associated with the biomarker of oxidative DNA damage, 8-hydroxy-2'-deoxyguanosine and increased susceptibility to smoking-related CAD.14,15-17
The high incidence of CAD in South Africans of Indian ancestry in Durban was reported in 1969.18 More than 40 years later, epidemiological data in South Africa (SA) showed Indians to have the highest mortality rates for CAD, followed by mixed race, white and black.19 Recent studies show a strong familial link with the history of diabetes mellitus type 2, hypertension and CAD, supporting a genetic basis for development.20,21
In light of the evidence that variants of GSTP1 and GSTM1 influence disease risk, we assessed the association of GST polymorphisms in young smoking and non-smoking South African Indian CAD patients.
Methods
A total of 102 young Indian CAD patients and 100 age-, race- and sex-matched controls were enrolled following institutional ethical approval (BE154/010). A full pathology report was compiled. The inclusion criterion for CAD patients were: Indian ancestry and unrelated, adults aged <45 years, and stable CAD confirmed at angiography. The exclusion criteria for controls included an acute coronary syndrome/revascularisation procedure in the preceding 3 months, chronic renal or liver disease, malignancy and known active inflammatory or infectious disease.
Genomic DNA was extracted from whole blood. Cells were transferred to 500 ìl lysis buffer (0.5% sodium dodecyl sulphate (SDS), 150 mM NaCl, 10 mM ethylenediaminetetra-acetic acid (EDTA), 10 mM Tris-HCl (pH 8.0)). RNase A (100 ìg/ml; DNase-free) was added and the mixture incubated (37°C, 1 h). Subsequently, proteinase K (200 ìg/ml) was added and incubated (3 h, 50°C) and a 0.1% volume 5 mM potassium acetate was added before centrifugation (5000 xg; 15 min). Supernatants containing genomic DNA were transferred to fresh tubes and extracted with 100% isopropanol and washed with 70% ethanol. DNA samples were solubilised in 10 mM Tris and 0.1 mM EDTA (pH 7.4, 4°C). DNA concentration was determined spectrophotometrically.
Differential polymerase chain reaction (PCR) was performed to assess the GSTM1 polymorphism: 268 base pair (bp) (â-globin) and 215 bp (GSTM1) PCR products were amplified using 30 pmol of primers for the GSTM1 gene and 10 pmol primers for the â-globin gene in a 25 ìl reaction (200 ìM of each deoxyribonucleotide (dNTP), 3.3 mM MgCl2, 1x Green GoTaq Flexi buffer, 1 U GoTaq DNA polymerase (Promega), 100 ng genomic DNA template). Primer sequences: GSTM1-Fwd 5'-GAACTCCCTGAAAAGCTAAAGC-3'; GSTM1-Rev 5'-GTTGGGCTCAAATATACGGTGG-3'; â-globin-Fwd 5'-CAACTTCATCCACGTTCACC-3'; â-globin-Rev 5'-GAAGAGCCAAGGACAGGTAC-3'. Following denaturation (96°C, 5 min), amplification was carried out with 25 cycles of denaturation (96°C, 30 sec), annealing (57°C, 30 sec) and extension (72°C, 30 sec), followed by a final extension (72°C, 5 min). Amplification products were electrophoresed on an agarose gel (4%, 0.5 mg/ml ethidium bromide) and visualised. The presence of a single 268 bp product was indicative of the homozygous null genotype. The amplification of a 215 bp product was indicative of the presence of the gene in either a homozygous positive or heterozygous state.
PCR-restriction fragment length polymorphism (PCR-RFLP) was used to determine the GSTP1 genotype. A 176 bp PCR product was amplified using 15 pmol of each primer (forward 5'-ACCCCAGGGCTCTATGGGAA-3' and reverse 5'-TGAGGGCACAAGAAGCCCCT-3') in a 25 ìl reaction containing 200 ìM of each dNTP, 2.5 mM MgCl2, 1x Green GoTaq Flexi buffer, 0.5U GoTaq DNA polymerase and 100 ng genomic DNA template. Following initial denaturation (96°C, 5 min), amplification was carried out with 30 cycles of denaturation (96°C, 30 sec), annealing (55°C, 30 sec) and extension (72°C, 30 sec). This was followed by a final extension (72°C, 5 min). Presence of the polymorphic restriction site was analysed by restriction endonuclease digestion of the PCR amplicon. Overnight digestion (37°C) was performed in 25 ìl reactions: 15 ìl PCR product, 4.5 ìl Buffer-R and 0.5 ìl (5 U) BsmAI (Fermentas). Amplicons homozygous for the G105 allele were completely digested and resulted in 2 restriction fragments (91 bp and 85 bp). Restriction fragments were electrophoresed on an agarose gel (3%, 0.5 mg/ml ethidium bromide) and visualised.
Statistical analyses were performed with GraphPad Prism software (version 5.0).
Results
Among the clinical parameters assessed between the groups, body mass index (BMI), hypertension, diabetes, triglycerides, fasting glucose and HbA1c were higher in patients than in controls (Table 1). Single nucleotide changes at codon 105 of the GSTP1 gene were investigated using RFLP-PCR. The genotype frequencies observed did not deviate from those predicted by Hardy-Weinberg statistics (GSTP1: p=0.294 in CAD patients, p=0.413 in controls; GSTM1 p=0.083 in CAD patients, p=0.64 in controls; chi-square test).
A significant skew towards frequency of the A105/A105 genotype was observed in CAD patients compared with the controls. In the control group, the homozygous A105/A105 and heterozygous A105/G105 genotypes were observed at frequencies of 48% and 45%, respectively. The frequency of the A105/A105 genotype was significantly higher in CAD patients (65% v. 48%, respectively; odds ratio (OR)=0.6011, 95% confidence interval (CI) 0.3803 - 0.9503, p=0.0377; Table 2). GSTM1 0/0 was also significantly more frequent in CAD patients (36% v. 18%; OR=2.593, p=0.0043; Table 3) compared with +/+ and +/0 genotypes.
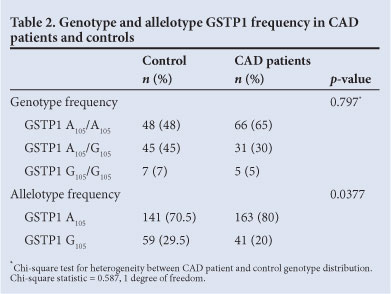

A higher frequency of the GSTP1 A105 and GSTM1 null alleles was observed in patients; therefore, we investigated whether there were genotypic differences in relative CAD risk in respect of these loci. Our findings suggest that the presence of either of these genotypes confers a significant CAD risk (Table 2). We stratified the groups according to smoking history: the number of smokers in the patient group was much higher than in the control, with a significant association between smoking and CAD (p<0.0001, OR=0.2245, 95% CI 0.1062 - 0.4746).
Most smokers presented with GSTP1 A105/A105 (p=0.0987, OR=0.5667, 95% CI 0.2952 - 1.088; Table 4) and GSTM1 0/0 (p=0.0221, OR=2.386 95% CI .137 - 5.009; Table 4). The higher relative risk of CAD associated with these genotypes is increased with smoking.
Discussion
The INTERHEART study ranked smoking as the second highest risk factor for myocardial infarction (OR=2.87, 99% CI).22 The Systemic Coronary Risk Evaluation project estimated the 10-year fatal cardiovascular risk to be twice as high for smokers v. non smokers for any given age, systolic BP and cholesterol level.23 The number of smokers worldwide is expected to reach 1.7 billion by 20 2 5.24,25 With current trends, smoking-related deaths are expected to rise from 4.8 million in 2000 to 8 million in 2030.25
Endothelial cell damage in vasculature is caused by changes in haemodynamics and oxidative stress, leading to thrombosis and atherosclerosis. The continuous cycle of inflammation from atherosclerotic plaques and arterial wall lesions contributes to elevated levels of C-reactive protein in CAD. The low-grade inflammation, recruitment and activation of leucocytes to the atherosclerotic lesion plays an important role in ROS generation in an attempt to circumvent the spread of foreign material, while simultaneously exposing biomolecules to oxidative stress.5
Our data show an association of wild-type GSTP1 A105/A105 and GSTM1 0/0 with CAD. The presence of allele GSTP1 G105 in the healthy control patients signals a possible role of this polymorphism in enhancing the efficacy of antioxidant mechanisms. GST acts as a general base catalyst, increasing the rate of GSH conjugation to hydrophobic substrates by deprotonation to GS- by an active tyrosinate. The change in codon 105 of the GST gene results in an amino acid substitution from Ile to Val. This was previously shown to cause deviations in the atomic co-ordinates of key H-site residue side-chains, thereby altering GST catalytic ability and increasing susceptibility to smoking-related CAD.15-17 Earlier literature, however, indicates 7 times more active conjugation to diol epoxides and increased catalytic efficiency to aromatic epoxides.8
GSTM1 0/0 results in complete absence of the enzyme isoform, which affects detoxification capacity. The strong association of this genotype in CAD patients indicates a key role in disease pathogenesis. The association between smoking and CAD in this cohort is in agreement with previous studies.15-17 The high percentage of patients who smoked and presented with GSTP1 A105/A105 and GSTM1 0/0 supports the hypothesis that GSTs play an important role in CAD.
Study limitations included sample size, lack of exclusion criteria based on body mass index (BMI), hypertension and diabetes.
Conclusion
This study provides evidence that the GSTP1 A105 allele and the GSTM1 null genotype are associated with CAD in young South Africans of Indian ancestry.
Acknowledgement. A Phulukdaree acknowledges an NRF-DAAD Scholarship.
References
1. McGill HC Jr, McMahan CA, Zieske AW, et al. Associations of coronary heart disease risk factors with the intermediate lesion of atherosclerosis in youth. Arterioscler Thromb Vasc Biol 2000;20:1998-2004. [http://dx.doi.org/10.1161/01.ATV.20.8.1998] [ Links ]
2. Blann AD, Kirkpatrick U, Devine C, Naser S, McCollum CN. The influence of acute smoking on leucocytes, platelets and the endothelium. Atherosclerosis 1998;141:133-139. [http://dx.doi.org/10.1016/S0021-9150(98)00163] [ Links ]
3. Nagy J, Demaster EG, Wittmann I, Shultz P, Raij L. Induction of endothelial cell injury by cigarette smoke. Endothelium 1997;5(4):251-263. [http://dx.doi.org/10.3109/10623329709052590] [ Links ]
4. Koul A, Bhatia V, Bansal MP. Effect of alpha-tocopherol on pulmonary antioxidant defence system and lipid peroxidation in cigarette smoke inhaling mice. BMC Biochem 2001;2:14. [http://dx.doi.org/10.1186/1471-2091-2-14] [ Links ]
5. Hulsmans M, Holvoet P. The vicious circle between oxidative stress and inflammation inatherosclerosis. J Cell Mol Med 2009;14:70-78. [http://dx.doi.org/10.1111/j.1582-4934.2009.00978.x] [ Links ]
6. Hayes JD, McLellan LI. Glutathione and glutathione-dependent enzymes represent a co-ordinately regulated defence against oxidative stress. Free Radic Res 1999; 31:273-300. [http://dx/doi. org/10.1080/10715769900300851] [ Links ]
7. Sies H. Glutathione and its role in cellular functions. Free Radic Biol Med 1999; 27:916-921. doi10.1016/S089-5849(99)00177-x [ Links ]
8. Hu X, Ji X, Srivastava SK, et aL Mechanism of differential catalytic efficiency of two polymorphic forms of human glutathione S-transferase P1-1 in the glutathione conjugation of carcinogenic diol epoxide of chrysene. Arch Biochem Biophys 1997;345:32-38. [http://dx.doi.org/10.1006/abbi.1997.0269] [ Links ]
9. Gong XG, Ji J, Xie J, Zhou Y, Zhang JY, Zhong WT. Expression, purification, and bioactivity of GST-fused v-Src from a bacterial expression system. J Zhejiang Univ Sci B 2006;7:13-19. [http://dx.doi.org/10.1631/jzus.2006.B0013] [ Links ]
10. Zanobetti A, Baccarelli A, Schwartz J. Gene-air pollution interaction and cardiovascular disease: a review. JPCAD 2011;53:344-352. [http://dx.doi.org/10.1016/j.pcad.2011.01.001] [ Links ]
11. Schwartz J, Park SK, O'Neill MS, et al. Glutathione-S-transferase M1, obesity, statins, and autonomic effects of particles: gene-by-drug-by-environment interaction. Am J Respir Crit Care Med 2005;172:1529-1533. [http://dx/doi.org/10.1164/rccm.200412-1698OC] [ Links ]
12. Chahine T, Baccarelli A, Litonjua A, et al. Particulate air pollution, oxidative stress genes, and heart rate variability in an elderly cohort. EHP 2007;115:1617-1622. [http://dx.doi.org/10.1289/ehp.10318] [ Links ]
13. Madrigano J, Baccarelli A, Wright RO, et al. Air pollution, obesity, genes and cellular adhesion molecules. Occup Environ Med 2010;67:312-317. [http://dx/doi.org/10.1136/oem.2009.046193] [ Links ]
14. Ren C, Vokonas PS, Suh H, Fang S, Christiani DC, Schwartz J. Effect modification of air pollution on Urinary 8-Hydroxy-2'-Deoxyguanosine by genotypes: an application of the multiple testing procedure to identify significant SNP interactions. Environ Health 2010;9:78. [http://dx.doi.org/10.1186/1476-069X-9-78] [ Links ]
15. Palme, CN, Young V, Ho M, Doney A, Belch JJ. Association of common variation in glutathione S-transferase genes with premature development of cardiovascular disease in patients with systemic sclerosis. Arthritis Rheum 2003;48:854-855. [http://dx.doi.org/10.1002/art.10955] [ Links ]
16. Park JH, El-Sohemy A, Cornelis MC, Kim HA, Kim SY, Bae SC. Glutathione S-transferase M1, T1, and P1 gene polymorphisms and carotid atherosclerosis in Korean patients with rheumatoid arthritis. Rheumatol Int 2004;24:157-163. [http://dx.doi.org/10.1007/s00296-003-0347-7] [ Links ]
17. Singh N, Sinha N, Kumar S, Pandey CM, Agrawal S. Glutathione S-transferase gene polymorphism as a susceptibility factor for acute myocardial infarction and smoking in the north Indian population. Cardiology 2011;118:16-21. [http://dx.doi.org/10.1159/000324066] [ Links ]
18. Wainwright J. Cardiovascular disease in the Asiatic (Indian) population of Durban. S Afr Med J 1969;43:136-138. [ Links ]
19. Norman R BD, Schneider M, Pieterse D, Groenwald P. Revised Burden of Disease Estimates for the comparative risk factor assessment, South Africa 2000. Cape Town: Medical Research Council 2006. [ Links ]
20. Ranjith N, Pegoraro RJ, Rom L, Lanning PA, Naidoo DP. Renin-angiotensin system and associated gene polymorphisms in myocardial infarction in young South African Indians. Cardiovasc J S Afr 2004;15:22-26. [ Links ]
21. Ranjith N, Pegoraro RJ, Naidoo DP. Demographic data and outcome of acute coronary syndrome in the South African Asian Indian population. Cardiovasc J S Afr 2005; 16:48-54. [ Links ]
22. Yusuf S, Hawken S, Ounpuu S, et al. Effect of potentially modifiable risk factors associated with myocardial infarction in 52 countries (the INTERHEART study): case-control study. Lancet 2004;364:937-952. [http://dx.doi.org/10.1016/S0140-6736(04)17018-9] [ Links ]
23. Conroy RM, Pyorala K, Fitzgerald AP, et al. Estimation of ten-year risk of fatal cardiovascular disease in Europe: the SCORE project. Eur Heart J 2003;24:987-1003. [http://dx.doi.org/10.1016/S0195-668X(03)00114-3] [ Links ]
24. Mathers CD, Loncar D. Projections of global mortality and burden of disease from 2002 to 2030. PLoS Med 2006;3:e442. [http://dx.doi.org/10.1371/journal.pmed.0030442] [ Links ]
25. World Health Organization (WHO). The World Health Report 2004: Changing history. Geneva: WHO, 2004. [ Links ]
Accepted 15 March 2012.
Corresponding author: A A Chuturgoon (chutur@ukzn.ac.za)


{kind=link}
{kind=link}