










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSAMJ: South African Medical Journal
On-line version ISSN 2078-5135
Print version ISSN 0256-9574
SAMJ, S. Afr. med. j. vol.102 n.6 Pretoria Jun. 2012
RESEARCH
The characteristics of juvenile myasthenia gravis among South Africans
J M HeckmannI; P HansenII; R van ToornIII E LubbeVI; E Janse van RensburgV; J WilmshurstVI
IFCP (Neurol), PhD. Division of Neurology, Department of Medicine, Groote Schuur Hospital and University of Cape Town
IIFCPaed, MB ChB. Department of Paediatrics, Chris Hani Baragwanath Hospital and University of the Witwatersrand, Johannesburg
IIIFCPaed, MB ChB. Department of Paediatrics, Tygerberg Children's Hospital and Stellenbosch University, Cape Town
IVFCPaed, MB ChB. Department of Paediatrics, Steve Biko Academic Hospital and University of Pretoria
VMMed (Neurol).Departments of Neurology and Paediatrics, Universitas Hospital and University of the Free State, Bloemfontein
VIFCP, MD. Departments of Paediatric Neurology and Neurophysiology, Red Cross War Memorial Children's Hospital and School of Child and Adolescent Health, University of Cape Town
ABSTRACT
OBJECTIVES: To report the characteristics of juvenile-onset (<20 years) myasthenia gravis (MG) in Africa.
SUBJECTS AND METHODS: Six South African centres collected data which included acetylcholine receptor-antibody (AChR-ab) status, delay before diagnosis, MG Foundation of America grade at onset, maximum severity and severity at last visit, therapies, outcomes and complications.
RESULTS: We report on 190 individuals with a 4-year median follow-up (interquartile range (IQR) 1 - 8). The median age at symptom onset was 7 years (IQR 4 - 14). Ocular MG (26%) occurred among younger children (mean 5.1 years) compared with those developing generalised MG (74%) (mean 10.2 years) (p=0.0004). Remissions were obtained in 45% of generalised and 50% of ocular MG patients, of whom the majority received immunosuppressive treatment, mainly prednisone. Children with post-pubertal onset had more severe MG, but deaths were infrequent. Thymectomies were performed in 43% of those with generalised MG who suffered greater maximum disease severity (p=0.002); there was a trend towards more remissions in the thymectomy group compared with the non-thymectomy group (p=0.057). There was no racial variation with respect to AChR-ab status, maximum severity, or use of immunosuppression. However, 23% of children of African genetic ancestry developed partial or complete ophthalmoplegia as a complication of generalised MG (p=0.002).
CONCLUSION: Younger children developed ocular MG and older children generalised MG. Persistent ophthalmoplegia developing as a MG complication is not uncommon among juveniles of African genetic ancestry. A successful approach to the management of this complication that causes significant morbidity is, as yet, unclear.
Juvenile myasthenia gravis (MG), defined as symptom onset before the age of 20, is generally less common than adult MG,1,2 and has been shown to have different sex and racial frequencies when white, African-American and Asian populations are compared.3 There are no data on juvenile MG from the African continent.
One centre has described racial differences in the outcome of generalised MG among South Africans, finding that severe treatment-resistant extra-ocular muscle (EOM) paresis or ophthalmoplegia occurred almost exclusively in subjects of African genetic ancestry.4 A higher proportion of individuals who developed partial or complete ophthalmoplegia presented with MG symptoms at a young age.4
We present the data from 190 subjects with juvenile MG collected from 6 academic centres throughout South Africa, report the frequency of the ophthalmoplegic complication in ocular and generalised MG and outcome in response to treatment.
Methods
The records of all patients up to June 2011 with a diagnosis of autoimmune MG from 6 tertiary care hospitals across South Africa were analysed: Groote Schuur Hospital (GSH), Cape Town; Red Cross War Memorial Children's Hospital (RCWMCH), Cape Town; Tygerberg Children's Hospital (TCH), Cape Town; Chris Hani Baragwanath Hospital (BARA), Soweto; Steve Biko Academic Hospital (SBA), Pretoria; Universitas Hospital (UH), Bloemfontein.
The diagnosis of MG was based on fatiguable weakness, a positive response to cholinesterase inhibitors and a positive acetylcholine receptor (AChR) antibody test (>0.2 nmol/l)1 or a positive response to immunotherapy such as plasma exchange or intravenous immunoglobulin (IVIg) or oral prednisone. Where the AChR antibody test was not performed or the result unknown, but there was a clear response to immunosuppression, the subject was classified as having either ocular (see later) or generalised MG.
Subjects with probable congenital myasthenic syndromes, i.e. fatiguable weakness but without any of the other criteria listed above, were excluded from the analyses: GSH (4), RCWMCH (1), TCH (0), BARA (1), SBA (0), UH (0). Neonates born to mothers with MG who showed transient symptoms (neonatal myasthenia) were not included.
The diagnosis of ocular MG was applied to those subjects with MG symptoms confined to the ocular muscles for the duration of their clinic attendance. We followed the MG Foundation of America (MGFA) Task Force recommendations5 (see footnote, Table 1) to determine clinical grade at onset and the 'post-intervention status' to categorise therapeutic response; where centres did not use the MGFA categorisation, minimal manifestations status (MMS) was used to identify patients 'in remission' or without symptoms, MMS-0 refers to those without treatment, MMS-1 refers to cholinesterase inhibitors (pyridostigmine) only, MMS-3 refers to subjects also undergoing immunosuppressive therapies. Immunosuppressive therapies consisted of prednisone with or without steroid-sparing agents, intravenous immunoglobulin (IVIg) or plasma exchange. Among those subjects undergoing thymectomy, the delay in months/ years between symptom onset and thymectomy was documented, as was thymic histology.
Subjects from Cape Town had been followed up for the longest period due to the referral system between the paediatric units of TCH and RCWMCH and the MG clinic at GSH. Furthermore, a prospective observational database was established at GSH in 1996 and updated 6-monthly, recording a quantified MG score on each patient, outcome and number of paralysed EOMs and treatment.4 In 2010 the other centres were co-opted into assessing their own patient records, largely retrospectively, and documenting age at onset, presenting MGFA grade, maximum severity, duration of follow-up, outcome on treatment and specifically paresis of EOMs and eyelids/ptosis. EOM paresis does not refer to the fatiguable weakness expected with MG but rather spontaneous and persistent pareses/ paralysis of one or more EOMs. If the EOMs could not be precisely examined and rated the investigators summarised these as partial (at least 6/12 (50%) of EOMs affected) or complete ophthalmoplegia (all 12 EOMs affected, invariably with bilateral ptosis).4
We have grouped patients according to race, predominantly by self-reporting (GSH) or mother tongue (remainder), as the ophthalmoplegic MG complication was previously described to show significant racial bias in a GSH cohort.4 The study protocol was approved by the ethics committees of all the participating universities.
Statistical analyses
Normally distributed data were presented as means and standard deviations (SD) and non-normally distributed data by medians and interquartile ranges (IQR). As appropriate, the Student t-test, Mann-Whitney test, chi-square test (or Fisher's exact test) were used to compare data. The Kruskal-Wallis one-way ANOVA was used to compare continuous variables not normally distributed for more than two groups, e.g. racial categories, and the Spearman rank-order correlation coefficient was used to assess relationships between ordinal variables between two groups. The MGFA and EOM grades were considered ordinal variables. The racial variation of EOMs or MGFA grades was assessed using the Kruskal-Wallis test. All analyses were two-sided and a p-value <0.05 was considered significant. Statistical analyses were done using STATISTICA 9 (Statsoft).
Results
The records of 190 juvenile MG patients who developed symptoms at a median age of 7 years (IQR 4 - 14, range 0.5 - 19.7) were assessed. The distribution of ages at onset showed two peaks corresponding to a pre-pubertal group (onset <12 years, N=126; 66%) with a mean age of 5.6 years (SD±2.9) and a post-pubertal group (N=64; 34%) with a mean age of 15.6 years (SD±2.2). Children with ocular MG presented at a younger age than those with generalised MG (p=0.0004) (Table 1). Subjects presenting with generalised symptoms were diagnosed after a longer delay than those presenting with ocular symptoms (mean 1.2±2.1 v. 0.8±1.4 years, respectively; p=0.16). Patients were followed up for a median period of 4 years (IQR 1 - 4, range 0.2 - 44).
Additional auto-immune disease was found in 12 (6%) individuals (1 patient had 2 auto-immune diseases), including systemic lupus erythematosus (2), insulin-dependent diabetes mellitus (2), juvenile rheumatoid arthritis (1), vitiligo (4), thyroid disease (2), psoriasis (1) and alopecia areata (1). Nine individuals had family members with an auto-immune disease of which MG (4% of cohort) was the commonest (the mothers of 2 individuals had MG and a further 3 had a second/third-degree MG relative with MG). There was 1 individual each with additional epilepsy, neurofibromatosis and HIV infection. No individuals developed cancer.
Ocular MG
Fifty patients had ocular disease as the sole manifestation of MG, with a mean age at symptom onset of 5.1 years (SD±3.5). Children of African or recent African ancestry showed a trend towards developing ocular MG rather than generalised MG (p=0.056) when compared with those of European ancestry. Ocular MG was more frequent (N=47; 94%) in the pre-pubertal onset group (<12 years) than in those with post-pubertal onset MG (N=3; 5%) (p<1x104). AChR antibody status was known in 42 of whom 26 (62%) were AChR antibody-positive. The median duration of follow-up was 1.5 years (IQR 0.8 - 4.0).
Forty-five per cent of the subjects were treated with cholinesterase inhibitors, while the remainder also received prednisone. The median time from symptom onset to initiating prednisone was 0.3 years (IQR 0.2 - 0.5). Remissions were obtained in 23 of 46 (50%), of whom 10 (20%) went into remission without using prednisone. Generally, remissions and functional improvement occurred among children of all races (p=0.17) and occurred irrespective of the use of immunosuppressive therapy (p=0.53).
Twenty-one (42%) of these children were followed up for >2 years during which time the MG symptoms remained active and confined to the ocular muscles. Of these subjects, 8 had persistent partial ophthalmoplegia (6 - 10 EOMs paresed) and 2 had complete ophthalmoplegia with bilateral ptosis; all were of African genetic ancestry (p=0.006).
Generalised MG
One hundred and forty subjects had generalised MG and presented with symptoms at an older age (mean 10.2 years, SD±5.4). Twenty-nine (21%) presented with ocular symptoms but developed generalised disease soon thereafter. Indigenous African children developed MG symptoms at a younger age than those with recent African and European genetic ancestry (p<1x104). The latency between symptom onset and diagnosis was on average 1.2 years (median 0.5; IQR 0.2 - 1.0). Of the 121 available AChR antibody test results, 98 (81%) were positive, irrespective of race (p=0.36). The median duration of follow-up was 5 years (IQR 2 - 10).
Overall, the median MGFA grade at disease presentation was mild (grade 2; IQR 2 - 3), deteriorating to a maximum grade of 3 or moderate disease severity (IQR 2 - 5) and improving to a final grade 1 (IQR 0 - 2), with 45% (60/133) in functional remission (minimal manifestation status). There was no racial difference in the MGFA grades at presentation or maximum severity but there was a difference in the final MGFA grade (p=0.044), likely a reflection of the persistent ophthalmopareses among children with African genetic ancestry (see later). Age at which symptoms started did not influence the final MGFA grade (p=0.81).
Disease onset was more frequent before onset of puberty (57%). Post-pubertal MG tended to be more severe than pre-pubertal MG (p=0.08), with a higher proportion of older children developing at least one MG crisis (p=0.017) (Table 2).

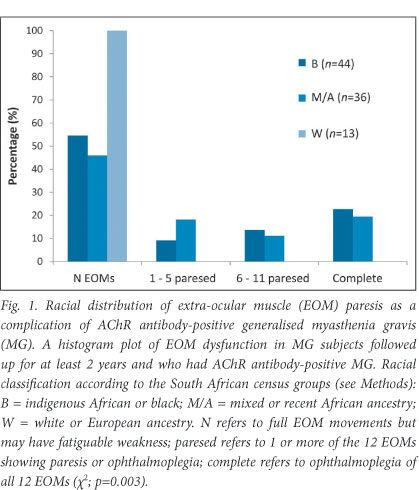
Partial or complete ophthalmoplegia developing as a complication of generalised MG was observed in 32 children (23%), all of whom were of either African or recent African genetic ancestry. Fig. 1 depicts the distribution of the number of extra-ocular muscles affected. The distribution of cases did not differ between the larger centres: combined Cape Town 27%; BARA 24%; combining SBA+UH 22% (p=0.69). The presence of MG-associated ophthalmoparesis correlated significantly with the race of the child (p=0.002), but not with gender (p=0.86), age at symptom onset (p=0.94), latency before diagnosis (p=0.60), use of immunosuppressive therapy (p=0.34), or years before the initiation of immunosuppressive therapy (p=0.70). Most of the patients (75%) were noted to have ophthalmoplegia at the time of MG diagnosis, but unlike the remaining MG manifestations the ophthalmoplegia remained treatment-resistant. The remaining 25% had some ocular MG manifestations at diagnosis, but only developed the complete ophthalmoplegia while being treated with immunosuppressive therapy.
Treatment and outcome
Immunosuppressive therapies were used in 83% of generalised MG cases, whereas 17% were treated symptomatically with pyridostigmine alone. The latency between diagnosis and initiation of immunosuppressive therapy was on average 1.7 years (median 0.5 years, IQR 0.2 - 2.0). Twelve individuals (10%) achieved remissions without immunosuppressive therapy; 3 received IVIg after initially presenting in MG crisis, but subsequently improved and did not need further immunosuppression. Sixty-nine subjects (63% of those on immunosuppression) were also treated with steroid-sparing agents (see Table 1). Four patients (3%) died of a myasthenic crisis, of whom 2 were not treated with immunosuppressive therapies. An additional 2 subjects died of unrelated causes - 1 of HIV/AIDS and 1 of ischaemic heart disease and arrhythmia, after 3 and 44 years of follow-up, respectively.
Of those with generalised MG, 43% underwent thymectomy, which was performed more frequently in older children (Table 2, p<1x104); half underwent surgery within 1 year of symptom onset. Thymic histological findings were available in 54: 36 (67%) had thymic hyperplasia, 3 (2%) had thymoma and 12 (22%) were normal. The patients undergoing thymectomy had greater disease severity than those who did not have surgery (p=0.002), and showed a trend towards proportionately more remissions achieved (55% v. 38%;p=0.057).
Owing to increased severity of disease, post-pubertal children were more likely to receive immunosuppressive therapies or to undergo thymectomy. However, the proportions obtaining remissions were similar (p=0.24).
The effect of thymectomy on the long-term remission rate in juvenile generalised MG
Analysis of the clinical outcome of 46 subjects (5 being lost to follow-up) diagnosed by one of three Cape Town clinics and followed up for a median of 7.5 years (range 0.3 - 44 years), shows that the thymectomy group had more severe disease (54% MGFA grade 4 or 5) compared with the non-thymectomy group (19% MGFA grade 4 or 5). Thymectomy did not influence the development of MG-associated ophthalmopareses (p=0.57) or frequency of remission (p=0.51). Twenty (of whom 16 had thymectomy) diagnosed with MG prior to 2001 still attend the clinic with active MG.
Discussion
We present the first description of an African juvenile MG cohort. Racial variation among juveniles with MG has been previously reported, with a very high incidence of ocular MG among Asian children.6 Interestingly, we show that children with African genetic ancestry, as opposed to European ancestry, also show a trend toward more ocular MG, with an age peak between 2 and 4 years.3 Moreover, we show that a substantial proportion of children with generalised AChR antibody-positive MG develop severe, treatment-resistant ophthalmoplegia associated with significant morbidity (Fig. 1).4 Apart from an isolated Canadian case (1 of 25)7 and 2 Korean children (2 of 24),8 an hitherto unrecognised MG complication of ophthalmoplegia and ptosis was described in a cross-sectional analysis of South Africans with generalised MG attending one centre (Cape Town).4 In that report, 13 patients with African genetic ancestry developed complete ophthalmoplegia and bilateral ptosis, 7 having developed MG symptoms in childhood.4 We now present data to show that either partial or complete ophthalmoplegia occurs among 23% of South Africans with juvenile MG attending different centres across South Africa. All subjects had African genetic ancestry (either indigenous African or recent African ancestry). The majority of these children had already developed either partial or complete ophthalmoplegia at the time of generalised MG symptom presentation, whereas the remainder developed it after treatment initiation. Research into the molecular basis9 and the optimal management of this complication is ongoing.
Juvenile MG accounts for less than 10 - 15% of all MG cases, with an incidence of 1 - 5 per million per year.3 The incidence of juvenile MG in South Africa is expected to be similar although under-recognition of MG is likely where access to specialist services is poor.1 As has been reported from the First World, pre-pubertal children tended to have more ocular presentations whereas post-pubertal cases were similar to adults with more generalised disease and female preponderance. The MGFA grades at presentation and frequency of AChR-positive generalised MG among this cohort were similar to other juvenile cohorts.3, 6,10 Remission rates in children are generally said to be high (11 - 37%).2,6,7,10 Our overall remission rate of 45%, of whom approximately 10% went into spontaneous remission, is comparable. Compared with generalised disease, ocular MG showed more frequent remissions and more often without the use of immunosuppressive therapies.
Thymectomy for AChR antibody-positive generalised MG is generally thought to be beneficial.3 Sixty of the generalised MG subjects, of whom the majority were post-pubertal children with more severe disease, underwent thymectomy; the histological examination showed the expected preponderance of thymic hyperplasia.6 A previous report which suggested that African-American children had a lower remission rate after thymectomy (=20%) compared with those of European ancestry (=58%)9 is not supported by our data, which comprised predominantly children with African genetic ancestry (in whom there was =45% remission). Although we showed a trend towards more remissions after thymectomy (p=0.057), a sub-analysis of the Cape Town cohort with longer patient follow-up did not reveal a difference in remission rate between those that did and those that did not have thymectomy (p=0.51; data not shown). However, the benefit of thymectomy versus no thymectomy is being studied for the first time in a prospective multinational controlled trial, the results of which are eagerly awaited.11
Generally the therapeutic approach in cases of generalised MG does not differ from that of adult-onset MG.3 Prednisone is first-line treatment in patients with symptoms who fail to respond to cholinesterase inhibitors (pyridostigmine).3,4 Azathioprine is the most commonly used steroid-sparing agent. While cyclosporine, mycophenolate mofetil or cyclophosphamide is second-line treatment elsewhere in the First World, we are increasingly using methotrexate, which has been shown to be cost-effective in adults with generalised MG12 and is frequently used in other juvenile auto-immune diseases.
Congenital myasthenic syndromes have been excluded from this analysis. Although ophthalmoplegia and ptosis may occur in congenital myasthenia they differ from acquired auto-immune MG described here, with the latter having circulating AChR antibodies and the non-ocular myasthenic manifestations responding significantly and appropriately to standard immunotherapy.
Although representing one of the largest published cohorts, there are a number of limitations to our study. Apart from Cape Town, data from other centres were retrospectively collected, albeit according to a study protocol. Further, the follow-up time was limited in many of the patients. The GSH cohort included fewer pre-pubertal patients, reflecting the fact that this hospital largely serves adults; the focus of the other five centres was on pre-pubertal subjects (Table 1). Nevertheless, the two larger centres (Cape Town and Soweto/BARA) reported similar frequencies of ophthalmoplegia.
In conclusion, the use of immunosuppression in juveniles with generalised MG not responding to pyridostigmine alone, is associated with a good remission rate.
Acknowledgements
The authors wish to acknowledge Dr Alvin Ndondo, paediatric neurologist at Red Cross War Memorial Children's Hospital, Professor Izelle Smuts, professor of paediatric neurology at Steve Biko Academic Hospital, and Dr Gail Sher, paediatric neurologist at Chris Hani Baragwanath Hospital, for assisting in the collection of clinical material used in the data analyses.
References
1. Bateman KJ, Schinkel M, Little F, Liebenberg L, Vincent A, Heckmann JM. Incidence of seropositive myasthenia gravis in Cape Town and South Africa. S Afr Med J 2007;97(10):959-962. [ Links ]
2. Rodriguez M, Gomez MR, Howard FM, Jr., Taylor WF. Myasthenia gravis in children: long-term follow-up. Ann Neurol 1983;13(5):504-510. [ Links ]
3. Evoli A. Acquired myasthenia gravis in childhood. Curr Opin Neurol 2010;23(5):536-540. [ Links ]
4. Heckmann JM, Owen EP, Little F. Myasthenia gravis in South Africans: racial differences in clinical manifestations. Neuromuscul Disord 2007;17(11-12):929-934. [ Links ]
5. Jaretzki A 3rd, Barohn RJ, Ernstoff RM, et al. Myasthenia gravis: recommendations for clinical research standards. Task Force of the Medical Scientific Advisory Board of the Myasthenia Gravis Foundation of America. Neurology 2000;55(1):16-23. [ Links ]
6. Chiang LM, Darras BT, Kang PB. Juvenile myasthenia gravis. Muscle Nerve 2009;39(4):423-431. [ Links ]
7. Mullaney P, Vajsar J, Smith R, Buncic JR. The natural history and ophthalmic involvement in childhood myasthenia gravis at the hospital for sick children. Ophthalmology 2000;107(3):504-510. [ Links ]
8. Kim JH, Hwang JM, Hwang YS, Kim KJ, Chae J. Childhood ocular myasthenia gravis. Ophthalmology 2003;110(7):1458-1462. [ Links ]
9. Heckmann JM, Uwimpuhwe H, Ballo R, Kaur M, Bajic VB, Prince S. A functional SNP in the regulatory region of the decay-accelerating factor gene associates with extraocular muscle pareses in myasthenia gravis. Genes Immun 2010;11:1-10. [http://dx.doi.org/10.1038/gene.2009.61] [ Links ]
10. Andrews PI, Massey JM, Howard JF jun., Sanders DB. Race, sex, and puberty influence onset, severity, and outcome in juvenile myasthenia gravis. Neurology 1994;44(7):1208-1214. [ Links ]
11. Aban IB, Wolfe GI, Cutter GR, et al. The MGTX experience: challenges in planning and executing an international, multicenter clinical trial. J Neuroimmunol 2008;201-202:80-84. [ Links ]
12. Heckmann JM, Rawoot A, Bateman K, Renison R, Badri M. A single-blinded trial of methotrexate versus azathioprine as steroid-sparing agents in generalized myasthenia gravis. BMC Neurol 2011;11:97. [http://dx.doi.org/10.1186/1471-2377-11-97] [ Links ]
Accepted 31 January 2012.
Corresponding author: J M Heckmann (jeanine.heckmann@uct.ac.za)


{kind=link}