










Servicios Personalizados
Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkSAMJ: South African Medical Journal
versión On-line ISSN 2078-5135
versión impresa ISSN 0256-9574
SAMJ, S. Afr. med. j. vol.102 no.6 Pretoria jun. 2012
RESEARCH
Burden of antituberculosis and antiretroviral drug-induced liver injury at a secondary hospital in South Africa
Charlotte SchutzI, IX; Zahiera IsmailVII; Charles John ProxenosVIII; Suzaan MaraisII, X; Rosie BurtonIII; Chris KenyonIV; Gary MaartensXV; Robert J WilkinsonV, XI, XIII, XVI; Graeme MeintjesVI, XII, XIV
IMB ChB, Dip HIV Man. Department of Medicine, Faculty of Health Sciences, University of Cape Town, and Infectious Diseases Unit, G F Jooste Hospital, Cape Town
IIMB ChB. Department of Medicine, Faculty of Health Sciences, University of Cape Town, and Infectious Diseases Unit, G F Jooste Hospital, Cape Town
IIIBSc, PhD, MB BS, MRCOG, FCP (SA), Cert ID Phys. Department of Medicine, Faculty of Health Sciences, University of Cape Town, and Infectious Diseases Unit, G F Jooste Hospital, Cape Town
IVMB ChB, MPH, MA, FCP (SA). Department of Medicine, Faculty of Health Sciences, University of Cape Town, and Infectious Diseases Unit, G F Jooste Hospital, Cape Town
VBM BCh, MA, DTM&H, PhD, FRCP (UK). Department of Medicine, Faculty of Health Sciences, University of Cape Town, and Infectious Diseases Unit, G F Jooste Hospital, Cape Town
VIMB ChB, FCP (SA), MRCP, PhD, Dip HIV Man. Department of Medicine, Faculty of Health Sciences, University of Cape Town, and Infectious Diseases Unit, G F Jooste Hospital, Cape Town
VIIMB ChB, Dip HIV Man. Infectious Diseases Unit, G F Jooste Hospital, Cape Town
VIIIMB ChB, Dip HIV Man. Infectious Diseases Unit, G F Jooste Hospital, Cape Town
IXMB ChB, Dip HIV Man. Clinical Infectious Diseases Research Initiative, Institute of Infectious Disease and Molecular Medicine, University of Cape Town
XMB ChB. Clinical Infectious Diseases Research Initiative, Institute of Infectious Disease and Molecular Medicine, University of Cape Town
XIBM BCh, MA, DTM&H, PhD, FRCP (UK). Clinical Infectious Diseases Research Initiative, Institute of Infectious Disease and Molecular Medicine, University of Cape Town
XIIMB ChB, FCP (SA), MRCP, PhD, Dip HIV Man. Clinical Infectious Diseases Research Initiative, Institute of Infectious Disease and Molecular Medicine, University of Cape Town
XIIIBM BCh, MA, DTM&H, PhD, FRCP (UK). Division of Medicine, Imperial College London, UK
XIVMB ChB, FCP (SA), MRCP, PhD, Dip HIV Man. Division of Medicine, Imperial College London, UK
XVMB ChB, MMed, FCP (SA), DTM&H. Division of Clinical Pharmacology, Faculty of Health Sciences, University of Cape Town
XVIBM BCh, MA, DTM&H, PhD, FRCP (UK). MRC National Institute for Medical Research, London, UK
ABSTRACT
BACKGROUND: G F Jooste Hospital (GFJH) is a secondary-level referral hospital in a high HIV and tuberculosis (TB) co-infection setting.
AIMS: To assess the proportion of significant drug-induced liver injury (DILI) due to tuberculosis treatment (TBT) and/or antiretroviral therapy (ART) among patients presenting with liver dysfunction at GFJH and to describe management and outcomes.
METHODS: A retrospective observational study was performed of all cases referred to GFJH with significant liver dysfunction from 1 January to 30 June 2009. Significant liver dysfunction was defined by alanine transaminase (ALT)=200 U/l or total bilirubin (TBR)=44 µmol/l. TBT- or ART-associated DILI was defined as significant liver dysfunction attributed to TBT and/or ART and which resulted in the halting of treatment or the adjustment thereof. Outcome measures included case numbers, descriptive data, and in-hospital and 3-month mortality.
RESULTS: A total of 318/354 cases of significant liver dysfunction were reviewed: 71 were classified as TBT- or ART-associated DILI, while liver dysfunction was attributed to other causes in the remainder. In-hospital and 3-month mortality of TBT- or ART-associated DILI patients was 27% (n=19) and 35% (n=25), respectively. The majority of deaths were related to sepsis or sepsis complicating liver dysfunction. Twenty-three patients (32%) were lost to follow-up; 23 (32%) were alive and in outpatient care 3 months after presentation.
CONCLUSIONS: TBT- or ART-associated DILI is a common reason for presentation at a referral hospital in South Africa. In-hospital and 3-month mortality are high. Prospective studies are needed to define optimal management.
An estimated 34 million people are HIV-infected globally; South Africa (SA) carries the highest burden with an estimated 5.6 million people infected.1 A major scale-up of public sector antiretroviral therapy (ART) has seen 1.4 million people start ART,2 and an increasing proportion of eligible individuals initiating treatment.3 Tuberculosis (TB) case notifications per annum in SA reached 396 554 in 2010, with 61% of incident TB cases HIV co-infected.4 TB prevalence is high at initiation of ART: 2 clinics in Cape Town reported concomitant TB treatment (TBT) in 25% and 40% of patients, respectively, at time of ART initiation.5,6 In SA, 42% of HIV-positive TB cases received both ART and TBT in 2009, compared with 18% in 2008.3
Drug-induced liver injury (DILI) is a well-recognised complication of TBT and ART. DILI is frequently encountered at healthcare facilities in SA and is especially challenging to manage in TB/HIV co-infected patients. Hepatotoxicity complicates TBT in5- 33% of patients.7 Studies reporting high rates include patients with subclinical elevations of liver enzymes. Depending on the regimen, hepatotoxicity develops in 9 - 30% of patients receiving ART.8 HIV infection itself is associated with an increased risk of major TBT side-effects.9 Liver dysfunction due to TBT or ART carries numerous risks, including: (i) direct morbidity and mortality due to liver failure, (ii) disease progression due to interruption of optimal therapy, (iii) complications of prolonged hospitalisation, and (iv) TBT or ART resistance related to interruptions.10
G F Jooste Hospital (GFJH), a secondary-level referral hospital in Cape Town, serves communities with a high burden of TB/HIV co-infection. There has been a major scale-up of treatment services for both infections in the hospital's catchment area in recent years. Patients from surrounding primary care HIV and TB clinics with adverse reactions to treatment of either disease, including significant symptoms or signs of liver injury, are referred for management to GFJH.
We aimed to assess the proportion of significant liver dysfunction caused by TBT and/or ART among patients with liver dysfunction presenting to a secondary hospital (GFJH), and to describe management and outcomes.
Methods
A retrospective observational study was conducted and results of all liver function tests (LFTs) performed at the GFJH National Health Laboratory Service laboratory from 1 January to 30 June 2009 were reviewed. Hepatocellular injury is characterised by a marked rise in serum transaminases, aspartate aminotransferase (AST) and alanine transaminase (ALT). Cholestatic liver injury is characterised by a rise in alkaline phosphatase, gamma-glutamyltransferase and/ or raised total bilirubin (TBR).11 Significant hepatocellular injury and cholestatic injury was defined as resulting in a Grade 3 or 4 elevation of ALT>200 U/l (>5 times the normal upper limit) and TBR>44 umol/l (>2.5 times the normal upper limit), respectively.12 Patients who fulfilled one or both criteria were included. Sepsis was documented when clinical presentation was compatible and the admitting team managed infection as the primary cause of illness. Hepatic encephalopathy was documented by the admitting team.
Patient records were reviewed and data recorded on a standardised form. DILI was attributed to TBT or ART if either regimen was interrupted or adjusted. Cases not classified as TBT- or ART-associated DILI were classified as 'liver dysfunction due to other causes'. This included: (i) patients receiving TBT and ART and diagnosed with hepatic TB-associated immune reconstitution inflammatory syndrome (TB-IRIS), (ii) patients presenting with untreated disseminated TB, (iii) patients receiving TBT and/or ART and presenting with liver injury with no subsequent change or discontinuation of TBT or ART, and (iv) patients diagnosed with alternative causes of liver dysfunction.
The University of Cape Town Human Research Ethics Committee approved the study (HREC ref: 522/2009). Statistical analyses were conducted with STATA 11.1 software (2009).
Management
Patients receiving TBT and ART were managed at primary care clinics according to national treatment guidelines.13,14 Patients with significant symptoms or signs suggestive of DILI were referred to GFJH. Patients were admitted or managed as outpatients depending on clinical severity and LFT results. GFJH has an infectious diseases referral clinic with capacity to manage stable DILI patients as outpatients.
In patients with TBT-associated DILI, a decision was made to cease treatment temporarily or to switch to alternative and less hepatotoxic treatment. Cessation of TBT was defined as discontinuation of all TBT for longer than a day. In patients whose treatment had been interrupted, a less hepatotoxic regimen, typically consisting of streptomycin, ethambutol and ofloxacin, was usually commenced once LFT results improved. Rifampicin and isoniazid was then 're-challenged' in a stepwise manner after LFT results normalised. Rifampicin or isoniazid was started at a low dose and increased to full dose over a few days with concurrent monitoring of ALT. This was followed by similar introduction of the second drug. Once full-dose rifampicin and isoniazid were re-introduced, certain of the less hepatotoxic drugs were discontinued. Optimal TBT was defined as rifampicin-based therapy.
Depending on the suspected cause and severity of ART-associated DILI, generally either: (i) ART was ceased, (ii) a single drug substitution was made, or (iii) more hepatotoxic ART (e.g. nevirapine) was interrupted and replaced within a few days (e.g. by efavirenz 5 - 7 days later), while less hepatotoxic ART (e.g. stavudine and lamivudine) was continued. Optimal ART was defined as triple ART medication from at least 2 classes.
Outcome assessment
Clinical management and outcome data were ascertained by review of patient records. In-hospital and 3-month (within 90 days of presentation) mortality were recorded, including the cause of death where possible. Only in-hospital mortality was recorded for patients with liver injury due to other causes. Three-month retention in care was also recorded. In the case of TBT and/or ART-associated DILI, the Clinicom and eKapa electronic databases were consulted to ascertain follow-up and mortality documented elsewhere in the Western Cape. Loss to follow-up was defined as inability to trace any patient data after discharge, within 3 months of presentation.
Results
A total of 354 patients met the criteria for inclusion; 36 (10.2%) were excluded due to incomplete or missing records. Of 318 cases reviewed, 71 were classified as TBT- and/or ART-associated DILI. In 247 cases, liver injury was attributed to other causes (Fig. 1). Among many other causes of liver injury, the most common was sepsis-induced liver dysfunction, evident from clinical presentation and LFT results; 27 patients receiving TBT or ART were not diagnosed with TBT- or ART-associated DILI because no alteration was made to TBT or ART.
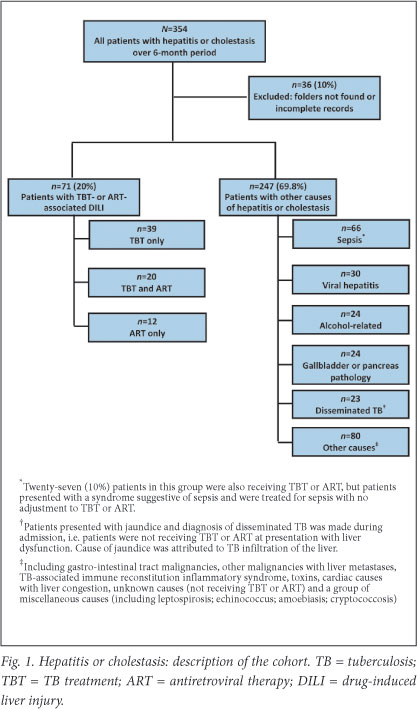
Baseline characteristics
Of the 318 patients, 47% were women and 41% were HIV-infected (Table 1). At presentation, 18% were receiving ART, 26% were receiving TBT and 10% were receiving concomitant TBT and ART. ART regimens included: (i) stavudine (or zidovudine), lamivudine and efavirenz (n=17; 53%); (ii) stavudine (or zidovudine), lamivudine and nevirapine (n=9; 28%); and (iii) protease inhibitor (PI)-based ART (n=4; 13%). Three patients receiving TBT were treated with PI-based ART and thus double-dose lopinavir/ritonavir. In the TBT- or ART-associated DILI category, a history of cotrimoxazole prophylaxis was available in 48/60 (80%) of the HIV-seropositive patients, and of these, 28 (47%) were receiving cotrimoxazole at presentation. Fifty-seven (80%) patients in this category were screened for hepatitis B surface antigen; 8 (14%) were positive.
Clinical presentation and management
Median length of hospital stay was 13 days (interquartile range (IQR) 7 - 20) in the 87% of TBT- or ART-associated DILI patients who were admitted. Patients presented as follows: 56 (79%) with cholestasis (TBR>44 ìmol/l), 31 (44%) with hepatocellular injury (ALT>200 U/l) and 18 (25%) with both. Fifteen (21%) had hepatic encephalopathy (Table 2). One or more possible alternate causes of liver dysfunction, including sepsis and TB-IRIS, were documented in 53 (75%) of patients.
TBT was ceased in 29 (49%) patients, with a median of 16.5 (IQR 14 - 26) days off optimal treatment. Less hepatotoxic TBT was initiated in 38 (64%) patients prior to re-challenge with rifampicin/isoniazid. Change in ART was individualised and varied widely.
Optimal rifampicin-based TBT was not re-introduced in 27/59 (45.8%) patients who presented while receiving TBT, for the following reasons: (i) death (n=14), (ii) transfer to another facility without particulars of subsequent re-challenge (n=5), (iii) absence of evidence for initial TB diagnosis (n=4), (iv) discharge with less hepatotoxic TBT and intention to re-challenge at a later stage (n=2), (v) presentation of DILI while receiving less hepatotoxic TBT which was continued while ART was adjusted (i.e. managed as ART-associated DILI) (n=1), (vi) completion of TBT (n=1), or (vii) patient absconded (n=1).
ART was interrupted in 11 patients (34%) and altered in 23 (74%). Patients spent a median of 25 (IQR 7 - 40) days off optimal ART (triple therapy). In 27 (38%) TBT- or ART-associated DILI patients, additional investigations were performed, including abdominal ultrasound (n=24), computed tomography (CT) of the abdomen (n=2), and liver biopsy (n=1; performed during DILI relapse).
Patients who were discharged and followed up as outpatients (n=21, 29.6%) had a median of 4.5 visits in the 3 months following presentation. Seven (9.9%) patients had subsequent relapse of hepatitis or cholestasis; 5 (71.4%) were re-admitted to hospital for a median of 12 days (IQR 12 - 21); 1 died in hospital due to sepsis and 6 were alive and in care 3 months after presentation of relapse.
Patient outcomes
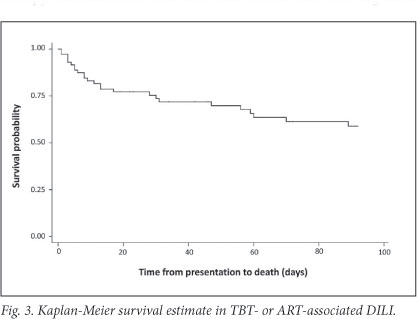
The Kaplan-Meier survival estimate is shown in Fig. 3. In-hospital mortality of the cohort was 27% (n=19) and 53% in patients who presented with hepatic encephalopathy. Common causes of death during initial admission included sepsis, a combination of sepsis and active TB or DILI, and liver failure (Fig. 2). Three of19 in-hospital deaths occurred after completion of TBT re-challenge; causes were sepsis (2) and unknown (1, unexpected death after initial improvement). At 3 months after presentation, 25 (35%) patients had died, 23 (32%) were alive and in care, and 23 (32%) were lost to follow-up (Table 3).
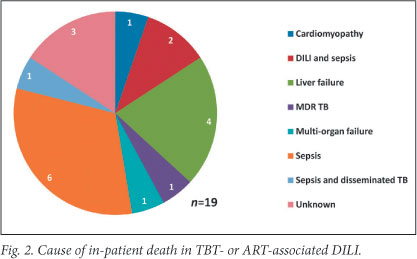
In cases of 'liver dysfunction due to other causes', 71 (28.7%) patients died during admission. This included 34 patients admitted with sepsis, of whom 14 were receiving TBT or ART.
Discussion
To our knowledge, this is the first study describing the management, outcome and high burden of TBT- or ART-associated DILI at a referral hospital in a high TB/HIV prevalence community. Over 6 months there were 71 cases. This may reflect under-ascertainment, accepting that -in the absence of a confirmatory test for DILI - some patients admitted with TBT- or ART-associated DILI would have been misclassified as having other causes of liver dysfunction such as TB-IRIS or sepsis.
TBT was a frequent cause of DILI in our study. TBT-attributed DILI may reduce TBT effectiveness due to a negative effect on adherence, treatment interruption and substitution with less effective TB medication. TBT-associated DILI may be fatal if not recognised and managed early.10
We attributed DILI to ART in 23/32 patients receiving such therapy. Medication of both first- and second-line ART regimens may cause DILI. Nucleoside reverse transcriptase inhibitors (NRTIs) may cause fatty liver related to mitochondrial toxicity. Non-nucleoside reverse transcriptase inhibitors (NNRTIs) cause liver injury related to immune-mediated hypersensitivity. The mechanism of PI-induced DILI is not entirely understood.8 In patients receiving lopinavir/ritonavir, additional ritonavir or double-dose lopinavir/ritonavir is required to counteract rifampicin induction, and high incidence of hepatotoxicity has been reported in such patients.15,16 Three patients were receiving lopinavir/ ritonavir-based ART and TBT; 2 were alive and in care 3 months after presentation and 1 was lost to follow-up.
Twenty patients were on concomitant rifampicin-based TBT and ART, which have overlapping toxicities. The cause of liver dysfunction in these patients is likely multi-factorial. The pathogenic mechanisms of DILI are complex, unpredictable and related to medication and genetic factors.11,17 Management is complex and needs to be individualised. TBT or ART was ceased or altered in all 20 cases, leading to prolonged hospital admission for drug re-challenge and increased vulnerability to disease progression and other infections. There are considerable risks associated with prolonged hospital stay in the context of advanced HIV infection, particularly nosocomial sepsis.18 We observed more cases of DILI related to TBT alone (n=39) than with ART alone (n=12) or concomitant ART and TBT (n=20), possibly reflecting that more patients were initiated on TBT than on ART in the hospital catchment area during the study period. Alternatively, TBT may be a more common cause of DILI.
Adding to the complexity, cotrimoxazole may also cause DILI; prophylactic treatment is often stopped during management of DILI, rendering patients vulnerable to opportunistic infections. Chronic hepatitis B is also an important co-factor in DILI; concomitant TBT significantly increases the risk of hepatotoxicity.19 Hepatitis B surface antigen was positive in a minority of cases in our study; 8/57 patients with TBT- or ART-associated DILI.
A striking finding was the high mortality associated with TBT- or ART-associated DILI (35% at 3 months after presentation) with a median time to death of 11 days (IQR 5 - 31). Mortality was likely under ascertained, as it is possible that some patients recorded as lost to follow-up really died. In comparison, mortality in a general cohort of patients with HIV-associated TB in our setting was only 8% during 6 months of TBT.20
In our study, the most common cause of death ascertained was sepsis, highlighting the vulnerability of patients with liver injury to bacterial and other infections. Close monitoring for community-and hospital-acquired bacterial infections and early diagnosis with appropriate antimicrobial treatment may improve outcome. Liver failure was the cause of death in only a minority of cases. Further studies are required to define reasons for the high mortality among DILI patients; this rate may reflect direct and indirect consequences of DILI, or that DILI complicates poor prognoses owing to other disease-related factors.
We acknowledge several limitations of our study. The use of herbal, traditional or over-the-counter medication, all of which may cause liver injury, was poorly documented. A retrospective review has many limitations: data capture was limited to documentation by attending clinicians and 10% of records were not found for review.
Conclusions
Liver injury due to TBT- or ART-associated DILI necessitating referral to hospital was common and associated with high mortality in our study. The cause of liver injury in HIV-TB co-infected patients is likely multi-factorial and is complex to manage. Prospective studies are urgently needed to investigate optimal management strategies and improve outcomes of such patients. The use of early invasive investigations (e.g. liver biopsy) in the diagnosis and management of liver injury requires investigation.
Acknowledgements
Sources of support: Perinatal HIV Research Unit, the US Agency for International Development, and the President's Emergency Plan for AIDS Relief (SM and CS), Wellcome Trust (RJW and GM, WT 081667, 084323 and 088316); a Fogarty International Center South Africa TB/AIDS Training Award (GM and CS, NIH/FIC 1U2RTW007373-01A1, U2RTW007373 ICORTA); a European Union Grant (RJW, SANTE/2005/105-061-102); and MRC (RJW, U1175.02.002.00014.01). The funders had no role in study design, data collection and analysis, decision to publish, or manuscript preparation.
References
1. UNAIDS. World Aids Day Report. Berlin/Geneva: UNAIDS, 2011. http://www.unaids.org/en/media/ unaids/contentassets/documents/unaidspublication/2011/JC2216_WorldAIDSday_report_2011_ en.pdf (accessed 3 January 2012). [ Links ]
2. Motsoaledi A. How we're re-engineering the health system. Johannesburg: Politicsweb, 2011. http://www.politicsweb.co.za/politicsweb/view/ politicsweb/en/page71656?oid=238984&sn=Detail&p id=71616 (accessed 14 November 2011). [ Links ]
3. UNAIDS Republic of South Africa Country Progress Report on the Declaration of Commitment on HIV/AIDS. Berlin/Geneva: UNAIDS, 2010. http://www.unaids.org/en/dataanalysis/monitoringcountryprogress/2010 progressreportssubmittedbycountries/ southafrica_2010_country_progress_report_ en.pdf (accessed 14 November 2011). [ Links ]
4. WHO. Global Tuberculosis Control 2011. Geneva: WHO, 2011. http://www.who.int/tb/publications/ global_report/2011/gtbr11_full.pdf (accessed 14 November 2011). [ Links ]
5. Lawn SD, Bekker LG, Middelkoop K, Myer L, Wood R. Impact of HIV infection on the epidemiology of tuberculosis in a peri-urban community in South Africa: the need for age-specific interventions. Clin Infect Dis 2006;42(7):1040-1047. [ Links ]
6. Boulle A, Van Cutsem G, Hilderbrand K, et al. Seven-year experience of a primary care antiretroviral treatment programme in Khayelitsha, South Africa. AIDS 2010;24(4):563-572. [ Links ]
7. Saukkonen JJ, Cohn DL, Jasmer RM, et al. An official ATS statement: hepatotoxicity of antituberculosis therapy. Am J Respir Crit Care Med 2006;174(8):935-952. [ Links ]
8. Montessori V, Press N, Harris M, Akagi L, Montaner JS. Adverse effects of antiretroviral therapy for HIV infection. CMAJ.2004;170(2):229-238. [ Links ]
9. Yee D, Valiquette C, Pelletier M, et al. Incidence of serious side effects from first-line antituberculosis drugs among patients treated for active tuberculosis. Am J Respir Crit Care Med 2003;167(11):1472-1477. [ Links ]
10. Tostmann A, Boeree MJ, Aarnoutse RE, et al. Antituberculosis drug-induced hepatotoxicity: concise up-to-date review. J Gastroenterol Hepatol. 2008;23(2):192-202. [ Links ]
11. Navarro VJ, Senior JR. Drug-related hepatotoxicity. N Engl J Med 2006;354(7):731-739. [ Links ]
12. DAIDS. Table for Grading Adult and Pediatric Adverse Events. Version 1.0. 2004. http://rsc.techres.com/Document/safetyandpharmacovigilance/Table_for_Grading _Severity_of_Adult_Pediatric_Adverse_Events.pdf (accessed 28 December 2011). [ Links ]
13. Department of Health (DoH). National Tuberculosis Management Guidelines 2008. Pretoria: DoH, 2008. http://www.sasohn.org.za/images/ TBGUIDELINES2008SFINALFORPRINTINGMARCH09.pdf (accessed 28 December 2011). [ Links ]
14. Department of Health. National Antiretroviral Treatment Guidelines. Johannesburg: Jacana, 2004. [ Links ]
15. Nijland HM, L'Homme RF, Rongen GA, et al. High incidence of adverse events in healthy volunteers receiving rifampicin and adjusted doses of lopinavir/ritonavir tablets. AIDS 2008;22(8):931-935. [ Links ]
16. Cohen K, Meintjes G. Management of individuals requiring antiretroviral therapy and TB treatment. Curr Opin HIV AIDS 2010;5(1):61-69. [ Links ]
17. Russmann S, Kullak-Ublick GA, Grattagliano I. Current concepts of mechanisms in drug-induced hepatotoxicity. Curr Med Chem 2009;16(23):3041-3053. [ Links ]
18. Pepper DJ, Rebe K, Morroni C, Wilkinson RJ, Meintjes G. Clinical deterioration during antitubercular treatment at a district hospital in South Africa: the importance of drug resistance and AIDS defining illnesses. PLoS One 2009;4(2):e4520. [ Links ]
19. Hoffmann CJ, Charalambous S, Thio CL, et al. Hepatotoxicity in an African antiretroviral therapy cohort: the effect of tuberculosis and hepatitis B. AIDS 2007;21(10):1301-1308. [ Links ]
20. Pepper DJ, Marais S, Wilkinson RJ, et al. Clinical deterioration during antituberculosis treatment in Africa: Incidence, causes and risk factors. BMC Infect Dis 2010;10:83. [ Links ]
Accepted 27 January 2012.
Corresponding author: C Schutz (charlottelouw@discoverymail.co.za)


{kind=link}
{kind=link}
{kind=link}