










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSAMJ: South African Medical Journal
On-line version ISSN 2078-5135
Print version ISSN 0256-9574
SAMJ, S. Afr. med. j. vol.102 n.6 Pretoria Jun. 2012
FORUM
ANALYSIS
Inflammatory pathways in cervical cancer - the University of Cape Town's contribution
Kurt J Sales; Arieh A Katz
Are principal investigators at the MRC Research Group for Receptor Biology, at the Institute of Infectious Disease and Molecular Medicine and the Division of Medical Biochemistry, Faculty of Health Sciences, University of Cape Town
ABSTRACT
Cervical cancer is the leading gynaecological malignancy in southern Africa. The main causal factor for development of the disease is infection of the cervix with human papillomavirus. It is a multi-step disease with several contributing co-factors including multiple sexual partners, a compromised immune system and cervical inflammation caused by infections with Chlamydia trachomatis or Neisseria gonorrhoeae. Inflammation involves extensive tissue remodelling events which are orchestrated by complex networks of cytokines, chemokines and bio-active lipids working across multiple cellular compartments to maintain tissue homeostasis. Many pathological disorders or diseases, including cervical cancer, are characterised by the exacerbated activation and maintenance of inflammatory pathways. In this review we highlight our findings pertaining to activation of inflammatory pathways in cervical cancers, addressing their potential role in pathological changes of the cervix and the significance of these findings for intervention strategies.
Cervical cancer is the second most common cancer in women worldwide and the leading gynaecological malignancy in women in Africa.1 In 2008 the International Agency for Research on Cancer estimated that 493 243 women are newly diagnosed with cervical cancer annually. Of these, more than 273 000 die each year.1,2 It is estimated that around 80 000 women, of whom 60 000 die each year, live in Africa. Because there are inadequate cancer registries in many African countries, it is likely that these figures are a gross under-representation.
Infection of the cervix with high-risk human papillomavirus (HPV) is regarded as the main causal factor in cervical cancer.3 There are more than 150 genotypes of HPV with around 40 known to infect the anogenital tract, giving rise to genital warts or neoplastic lesions.3 Recently, 2 prophylactic vaccines against the main high-risk HPV variants, 16 and 18, have been introduced by Merck & Company (Gardasil) and GlaxoSmithKline (Cervarix). These induce an immune response that blocks initial HPV infection and confers protection against cancer associated with HPV 16 and 18 and some closely related variants. These have limited benefit for women already infected with high-risk HPV and in addition are out of reach of the majority of women in Africa due to the high costs involved. Although HPV infection initiates disease, cervical cancer is a multi-step process, with other contributing factors, including multiple sexual partners, tobacco carcinogens, a weakened immune system and sexually transmitted infection by human immunodeficiency virus (HIV), Chlamydia trachomatis and Neisseria gonorrhoeae, thought to contribute to the aetiology.
HPV initially infects basal keratinocytes and epithelial cells and uses the host's cellular machinery for replication and persistence.4 The HPV genome consists of 3 domains, a non-coding upstream region, an early region containing open reading frames E1, E2, E4, E5, E6 and E7, and a late region encoding the major and minor capsid proteins.4 In the vast majority of women, infections and HPV-induced lesions are transient and are naturally resolved. However, approximately 10 - 20% of women fail to eliminate the virus.4 In these cases, persistence of infection, viral integration and activation of inflammatory pathways have been linked to neoplastic transformation and malignant progression.4,5 In this review, we highlight our findings relating to the activation of inflammatory pathways in cervical cancers and address their role in disease progression.
Persistent HPV infection and inflammation
By broad definition, inflammation involves tissue remodelling events brought about by alterations to epithelial, vascular and immune cell function. These are orchestrated by specific molecular pathways involving a host of cytokines, chemokines, growth factors and lipid mediators.6 Compelling evidence has shown that the majority of cancers arise from sites of chronic irritation, infection and inflammation,7 solidifying the concept that chronic unabated inflammation is critical for tumour progression.
Persistent HPV infection and integration of E6 and E7 oncogenes into the host genome is considered key to development of cervical cancer.4 The HPV E6 and E7 early genes encode oncoproteins responsible for cervical neoplastic transformation3 by inactivating tumour suppressors as well as promoting the accumulation of genetic mutations.3,4 Although E6 and E7 oncogenes appear to be the main HPV genes involved in transformation, recent studies have highlighted an important role for E5 oncogene in tumorigenesis and immune cell modulation8 and regulation of late viral functions together with the E4 oncogene. In addition, E1 and E2 oncogenes encode replication factors and are thought to play a role in HPV persistence by allowing episomal copies of the virus to be maintained in the nucleus and partitioned into daughter cells during mitosis.9
Immune evasion is an essential aspect of HPV persistence and development of cervical cancer. Since there is no viraemia or cytolysis associated with initial viral infection of the cervix, there is no activation of the innate immune system and no inflammation. Despite this, the virus actively induces mechanisms to evade immune detection and ensure its success by deregulating the interferon pathway4 and via the down-regulation of pattern recognition receptors such as Toll-like receptor 9, thereby allowing infection to proceed undetected.4 The virus requires actively dividing cells and active host cellular machinery for replication and persistence.4 Once established, persistent infections promote alterations in the release of inflammatory cytokines which in turn can alter immune cell infiltration and inflammation. Alterations in immune responsiveness and elevated systemic levels of inflammatory cytokines have been observed in older women (about 50 years of age) with persistent HPV infection.5 Since this is the age group most likely to present with cervical cancer in the clinic, it is likely that sustained elevation in systemic cytokine release contributes to HPV-mediated tumorigenesis.
Although the direct association between HPV infection and inflammation is controversial, transgenic mouse models, expressing the early genes from HPV 16 under the control of the human keratin 14 promoter, have shown that HPV-induced lesions release the chemokine CCL2 which enhances macrophage recruitment into tumours via CCR2.10 In human neoplastic cervical epithelial cells, HPV 16 E5, E6 and E7 oncogenes have been shown to induce the inflammatory cyclo-oxygenase (COX)-prostaglandin axis, by elevating expression of the immediate early oncogene COX-2.11 These findings provide a direct link between HPV oncogenes and activation of potent inflammatory cascades, with known roles in promoting cancer. Thus, although HPV is not associated with inflammation at the initial point of infection, it is likely that following integration and transformation, persistent HPV infection drives inflammatory pathways, such as the COX-prostaglandin pathway in neoplastic epithelial cells, to promote immune cell infiltration, inflammation and tumour progression.
The inflammatory cyclo-oxygenase-prostaglandin pathway
COX enzymes, of which there are 2 isoforms in humans (COX-1 and COX-2), catalyse the rate-limiting conversion of arachidonic acid to the unstable intermediate prostaglandin H2, which in turn is converted by terminal prostaglandin synthase enzymes to specific classes of prostaglandins.12 For many years COX-1 was considered to be constitutively expressed in tissues at low levels, generating prostaglandins for normal physiological functions, whereas COX-2 was considered to be an immediate early gene involved in pathology.12 Studying tissue biopsies, we showed that COX-1 and COX-2 expression were both significantly elevated in the neoplastic epithelial and vascular endothelial cells of cervical cancers of all grades and stages.13,14 These findings highlighted a role for both COX isoforms in pathology of the cervix. In order to elucidate the role of COX-1 in cervical cancers, we used an in vitro model system where we stably expressed the COX-1 gene in cervical adenocarcinoma (HeLa) cells under the control of a tetracycline-inducible promoter (HeLa COX-1 TET-OFF system).14 Induction of COX-1 expression in HeLa cells caused a rapid and sustained elevation in the expression of COX-2 and terminal PGE synthase (PTGES), resulting in the biosynthesis of PGE2.14 Furthermore, the PGE2 was produced by both COX-1 and COX-2, indicating that they can contribute equally, or work synergistically, to promote cervical cancer.
The selectivity for prostaglandin production is determined by the terminal prostaglandin synthase enzyme present in cells expressing COX-1 and COX-2.12 Santin and colleagues15 showed that the terminal PTGES enzyme, which converts PGH2 to PGE2, is significantly over-represented in invasive cervical cancers. This is consistent with our observations of elevated biosynthesis of PGE2 in cervical cancers,13 suggesting a dominance of this prostaglandin in cervical cancer. PGE2 exerts its biological role via 4 subtypes of E-series prostanoid G protein-coupled receptors (PTGER1-4).14 These receptors are often co-expressed on the same cell. We found that cervical cancers expressed elevated PTGER2 and PTGER4 in addition to elevated expression of COX enzymes and biosynthesis of PGE2. Until recently, the molecular mechanisms regulating prostaglandin receptor expression in cervical cancer cells were unknown. However, in vitro studies have shown that HPV oncogenes and PGE2 can regulate the expression of prostaglandin receptors. For example, the HPV 16 E5 oncogene has been shown to regulate expression of PTGER4 in a cervical cancer cell line in a PGE2-cAMP-dependent manner.8 We have shown that PGE2, either directly16 or following induction of COX-1 and COX-2 in HeLa cells, using the HeLa COX-1 TET-OFF system, can regulate prostaglandin receptor (PTGER2/PTGER4) expression.14 These findings suggest that in cervical cancers the elevated PGE2 could regulate neoplastic cervical cell function in an autocrine/paracrine manner via the elevated PTGER2 and PTGER4 receptors. Indeed, our studies using tissue biopsies showed that cAMP levels were augmented in cervical cancer biopsies, relative to normal cervix, treated ex vivo with PGE2.13
Taken together, our findings demonstrate that following HPV infection and viral integration in cervical epithelial cells, activation of viral oncogenes induces COX enzyme expression, PGE2 biosynthesis and PTGER expression. In turn, PGE2 via PTGER can regulate tumour cell function via cAMP signalling.
Regulation of vascular function and immune cell recruitment by the COX-prostaglandin pathway
In several in vitro and in vivo model systems employing cell lines and rodents, overexpression of PGE2 as a consequence of elevated COX enzyme expression has been shown to promote tumorigenesis. This occurs by inducing tissue remodelling within the tumour by inhibiting apoptosis, enhancing cellular proliferation, facilitating tumour metastases and elevating angiogenesis.12 We have shown that PGE2, either directly, or biosynthesised following induction of COX-1 and COX-2 in HeLa cells, elevates the expression of potent pro-angiogenic factors such as basic fibroblast growth factor 2, vascular endothelial growth factor (VEGF) and angiopoietins.14,17 Following their biosynthesis and release from neoplastic cervical epithelial cells, angiogenic factors can then exert a paracrine activity on endothelial cells to enhance blood supply to facilitate tumour growth, as well as alter vascular permeability to allow extravasation of leucocytes and macrophages into the surrounding tissues.6
Macrophage infiltration into cervical tumours has been positively correlated with tumour vascularity18 and women with advanced-stage invasive cancer have higher blood neutrophil counts than those with early-stage disease.19 Although the precise mechanism for immune cell recruitment into the cervix in humans has not been elucidated, prostaglandins biosynthesised by COX enzymes in epithelial, stromal and vascular cells have been shown to induce the expression of a host of cytokines and chemokines. These can in turn act in an autocrine or paracrine manner in the cervix to enhance inflammation by promoting tissue remodelling and recruitment of immune cells via chemotaxis and extravasation, which in turn can promote disease progression.6
In order to allow for leucocyte extravasation, changes in the vasculature and angiogenesis are required. This involves tissue remodelling of the extracellular matrix, a process facilitated by matrix metalloproteinases (MMPs).6 Several studies have correlated transcription of HPV E6 and E7 with transcription of MMPs,20 suggesting that HPV oncogenes can drive tissue and vascular remodelling. Indeed, micro-array analysis has shown that HPV 16 E6 oncoprotein regulates several genes involved in tissue differentiation and remodelling, which are important for inflammation and tumour progression.21 Whether HPV oncogenes directly regulate these genes involved in tissue remodelling events, or drive their transcription via intermediary pathways such as the COX-prostaglandin pathway, remains to be determined.
Nonetheless, our studies, and others, highlight a mechanism whereby activation of a chronic inflammatory pathway following HPV infection and cellular transformation can induce tissue remodelling events in cervical epithelial cells. Disease progression is promoted by altered vascular function and angiogenesis via the increased biosynthesis and signalling of PGE2.
Seminal fluid as a regulator of cervical inflammation and cancer
The main route of HPV transmission is via exposure of the cervix to virus present in seminal fluid and in the infected partner's skin during coitus. In addition to being a vehicle for the dissemination of HPV, seminal fluid contains a diversity of molecules that include cytokines, angiogenic factors, proteases, protein kinases, transporter proteins, structural molecules and immune response proteins.22 Based on our research, we have proposed that the inflammatory environment of cervical cancers can be further modulated by these mediators present in seminal fluid.16,17 Deposition of seminal fluid into the female reproductive tract elicits a wave of cytokine release and recruitment and activation of leucocytes.23 Little is known about the effect of seminal fluid on the neoplastic cervical epithelium. However, it can promote the release of MMPs which can alter the integrity of the epithelial barrier at the endocervical canal and can enhance metastases24 and promote the release of local pro-inflammatory mediators to regulate immune cell recruitment.23
We have shown that seminal plasma can induce expression of COX-1 and COX-2 and the E-series prostaglandin receptors (PTGER1, PTGER3 and PTGER4) in normal cervical tissue explants (Fig. 1A) and neoplastic cervical epithelial cells.16 Furthermore, in addition to the inflammatory COX-prostaglandin receptor axis, seminal plasma induces the expression of inflammatory cytokine interleukin (IL)-6, chemokines (IL-8 and growth-regulated oncogene (GRO) alpha) and VEGF in cervical tissue explants (Fig. 1B). These observations have been confirmed by Sharkey et al.,23 who have shown that seminal fluid induces an inflammatory response in the cervix in humans after coitus, characterised by the influx of leucocytes and dendritic cells into the epithelium and stromal compartments and an accompanying increase in inflammatory cytokines such as IL-6 and IL-8. These data provide robust evidence for a regulatory role of seminal fluid on the cervical micro-environment in favour of inflammation which might facilitate disease progression.
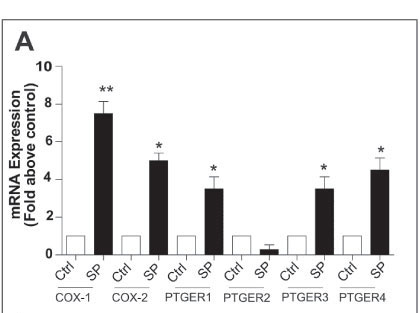
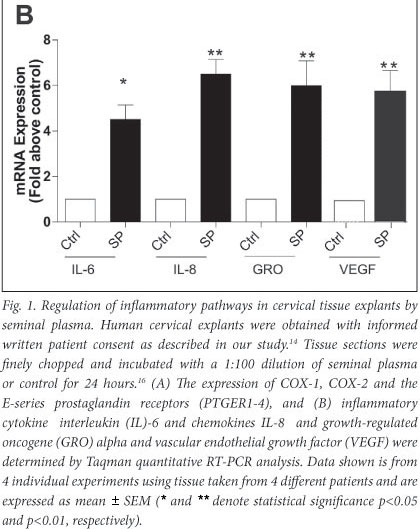
We earlier discussed the role of PGE2, produced by elevated COX enzyme expression in cervical cancers. PGE2 is abundant in seminal fluid, present at concentrations of up to 10 000-fold greater than at the site of chronic inflammation. We have shown that the PGE2 in seminal fluid can enhance the biosynthesis and release of VEGF from cervical cancer cells via the PTGER4-mediated transactivation of the epidermal growth factor receptor and extracellular signal-regulated kinase signalling pathways.17 The elevated synthesis and release of VEGF in turn can regulate vascular permeability to facilitate extravasation of immune cells from the vasculature into the tumour, as well as promote angiogenesis in cervical cancers.17 Taken together, our observations, as outlined in Fig. 2, suggest that repeated exposure of neoplastic cervical epithelial cells to seminal fluid can promote tissue remodelling events associated with inflammation. These exogenous inflammatory stimuli can act together with inflammatory stimuli, regulated endogenously by HPV oncogenes and COX enzymes, to augment cervical cancer progression.
Therapeutic management strategies
In Africa a large proportion of women have HPV infections; the majority of women with cervical cancer present with advanced-stage disease and poor prognosis. Treatment of early-stage cervical cancer is generally surgical, often combined with radiation and/ or chemotherapy. However, radiation and chemotherapy are not available in all African countries and it is evident that adequate national screening programmes to detect HPV and early cervical cancer precursors are needed.
In a number of tumour model systems, including colon cancer cells implanted into nude mice and carcinogen-induced tumours in rats, the application of non-steroidal anti-inflammatory drugs (NSAIDs) and selective COX enzyme inhibitors exhibit dramatic anti-cancer activity.12 This is mediated partially by reducing PGE synthesis in the COX-2-overexpressing cells, which in turn down-regulates the survival, metastatic, and angiogenic potentials of the cancerous tissue.12 Our observations of elevated biosynthesis and signalling of PGE2 in cervical cancers prompt us to suggest that inhibition of PGE2 secretion by the application of COX enzyme inhibitors may suppress growth and invasiveness of cervical carcinomas. One of the most widely available and cheapest NSAIDs is aspirin. Recent clinical trials have shown that long-term aspirin treatment can be beneficial in colorectal cancer.25 It is tempting to speculate that such anti-inflammatory agents may similarly prove beneficial and cost-effective for preventing progression of cervical cancer.
Our observations of the role of seminal plasma in regulating potent inflammatory and angiogenic pathways in neoplastic cervical epithelial cells suggest use of barrier contraceptives as a method of preventing disease, not only as a barrier against HPV transmission, but as a method of preventing the inflammatory actions of seminal fluid on the neoplastic cervical micro-environment. In the absence of barrier contraceptives, our research has highlighted the potential advantages of using prostaglandin receptor antagonists to prevent the activation and signalling of prostaglandin receptors by PGE2 present in seminal fluid. Potentially, these antagonists could be utilised in combination therapy with COX enzyme inhibitors such as aspirin to prevent endogenous production of PGE2 in the cervical tumour as well as the exogenous actions of prostaglandin present in the seminal fluid.
References
1. Arbyn M, Castellsague X, de Sanjose S, et al Worldwide burden of cervical cancer in 2008. Ann Oncol 2011;22(12):2675-2686. [ Links ]
2. Ferlay J, Shin HR, Bray F, et al Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer 2010;127(12):2893-2917. [ Links ]
3. zur Hausen H. Papillomaviruses in the causation of human cancers - a brief historical account. Virology 2009;384:260-265. [ Links ]
4. Stanley MA, Pett MR, Coleman N. HPV: from infection to cancer. Biochem Soc Trans 2007;35:1456-1460. [ Links ]
5. Kemp TJ, Hildesheim A, Garcia-Pineres A, et al. Elevated systemic levels of inflammatory cytokines in older women with persistent cervical human papillomavirus infection. Cancer Epidemiol Biomarkers Prev 2010;19:1954-1959. [ Links ]
6. Jabbour HN, Sales KJ, Catalano RD, Norman JE. Inflammatory pathways in female reproductive health and disease. Reproduction 2009;138:903-919. [ Links ]
7. Coussens LM, Werb Z. Inflammation and cancer. Nature 2002;420:860-867. [ Links ]
8. Oh JM, Kim SH, Lee YI, et al. Human papillomavirus E5 protein induces expression of the EP4 subtype of prostaglandin E2 receptor in cyclic AMP response element-dependent pathways in cervical cancer cells. Carcinogenesis 2009;30:141-149. [ Links ]
9. Bodily J, Laimins LA. Persistence of human papillomavirus infection: keys to malignant progression. Trends Microbiol 2011;19:33-39. [ Links ]
10. Pahler JC, Tazzyman S, Erez N, et al. Plasticity in tumor-promoting inflammation: impairment of macrophage recruitment evokes a compensatory neutrophil response. Neoplasia 2008;10:329-340. [ Links ]
11. Subbaramaiah K, Dannenberg AJ. Cyclooxygenase-2 transcription is regulated by human papillomavirus 16 E6 and E7 oncoproteins: evidence of a corepressor/ coactivator exchange. Cancer Res 2007;67:3976-3985. [ Links ]
12. Sales KJ, Jabbour HN. Cyclooxygenase enzymes and prostaglandins in reproductive tract physiology and pathology. Prostaglandins Other Lipid Mediat 2003;71:97-117. [ Links ]
13. Sales KJ, Katz AA, Davis M, et al. Cyclooxygenase-2 expression and prostaglandin E(2) synthesis are up-regulated in carcinomas of the cervix: a possible autocrine/paracrine regulation of neoplastic cell function via EP2/EP4 receptors. J Clin Endocrinol Metab 2001; 86:2243-2249. [ Links ]
14. Sales KJ, Katz AA, Howard B, et al. Cyclooxygenase-1 is up-regulated in cervical carcinomas: autocrine/paracrine regulation of cyclooxygenase-2, prostaglandin e receptors, and angiogenic factors by cyclooxygenase-1. Cancer Res 2002;62:424-432. [ Links ]
15. Santin AD, Zhan F, Bignotti E, et al. Gene expression profiles of primary HPV16- and HPV18-infected early stage cervical cancers and normal cervical epithelium: identification of novel candidate molecular markers for cervical cancer diagnosis and therapy. Virology 2005;331:269-291. [ Links ]
16. Sales KJ, Katz AA, Millar RP, Jabbour HN. Seminal plasma activates cyclooxygenase-2 and prostaglandin E2 receptor expression and signalling in cervical adenocarcinoma cells. Mol Hum Reprod 2002;8:1065-1070. [ Links ]
17. Muller M, Sales KJ, Katz AA, Jabbour HN. Seminal plasma promotes the expression of tumorigenic and angiogenic genes in cervical adenocarcinoma cells via the E-series prostanoid 4 receptor. Endocrinology 2006;147:3356-3365. [ Links ]
18. Mazibrada J, Ritta M, Mondini M, et al. Interaction between inflammation and angiogenesis during different stages of cervical carcinogenesis. Gynecol Oncol 2008;108:112-120. [ Links ]
19. Tavares-Murta BM, Mendonca MA, Duarte NL, et al. Systemic leukocyte alterations are associated with invasive uterine cervical cancer. Int J Gynecol Cancer 2010;20:1154-1159. [ Links ]
20. da Silva Cardeal LB, Brohem CA, Correa TC, et al. Higher expression and activity of metalloproteinases in human cervical carcinoma cell lines is associated with HPV presence. Biochem Cell Biol 2006;84:713-719. [ Links ]
21. Duffy CL, Phillips SL, Klingelhutz AJ. Microarray analysis identifies differentiation-associated genes regulated by human papillomavirus type 16 E6. Virology 2003;314:196-205. [ Links ]
22. Fung KY, Glode LM, Green S, Duncan MW. A comprehensive characterization of the peptide and protein constituents of human seminal fluid. Prostate 2004;61:171-181. [ Links ]
23. Sharkey DJ, Tremellen KP, Jasper MJ, Gemzell-Danielsson, Robertson SA. Seminal fluid induces leukocyte recruitment and cytokine and chemokine mRNA expression in the human cervix after coitus. J Immunol 2012 (in press). [ Links ]
24. Jeremias J, Witkin SS. Effect of human seminal fluid on production of messenger ribonucleic acid for metalloproteinase 2 and metalloproteinase 9 in cervical epithelial carcinoma cells. Am J Obstet Gynecol 1999;181:591-595. [ Links ]
25. Burn J, Gerdes AM, Macrae F, et al. Long-term effect of aspirin on cancer risk in carriers of hereditary colorectal cancer: an analysis from the CAPP2 randomised controlled trial. Lancet 2011;378(9809):2081-2087. [ Links ]
Accepted 23 March 2012.
Corresponding author: K J Sales (kurt.sales@uct.ac.za)


{kind=link}