










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSAMJ: South African Medical Journal
On-line version ISSN 2078-5135
Print version ISSN 0256-9574
SAMJ, S. Afr. med. j. vol.102 n.6 Pretoria Jun. 2012
FORUM
ANALYSIS
Polyglutamine disease: From pathogenesis to therapy
Lauren M WatsonI; Janine ScholefieldII; L Jacquie GreenbergIII; Matthew J A WoodIV
ILauren Watson is a PhD student and member of the Neurodegenerative Disorders research group, Division of Human Genetics, Faculty of Health Sciences (FHS), University of Cape Town (UCT). At the time of writing she was a visiting Commonwealth Scholar at the University of Oxford
IIJanine Scholefield obtained her PhD in the Division of Human Genetics, UCT. At the time of writing she was a Nuffield Medical Fellow at the University of Oxford. She is currently a visiting post-doctoral fellow at the Council for Scientific and Industrial Research (CSIR) in South Africa
IIIProfessor Jacquie Greenberg has worked at UCTs FHS since 1971 and obtained her doctorate in 1990. She is a medical scientist and genetic counsellor, and heads up the molecular genetics diagnostic laboratory at UCT/National Health Laboratory Service. She is the principal investigator of the Neurodegenerative Disorders research group
IVMatthew Wood is Professor of Neuroscience at the University of Oxford, and Fellow and Tutor in Medicine at Somerville College, Oxford. He is an Honorary Professor at UCT. He directs a leading research group investigatingRNA-based therapies for neurological and neuromuscular diseases
ABSTRACT
Polyglutamine diseases are inherited neurodegenerative conditions arising from expanded trinucleotide CAG repeats in the diseasecausing gene, which are translated into polyglutamine tracts in the resultant protein. Although these diseases share a common type of mutation, emerging evidence suggests that pathogenesis is complex, involving disruption of key cellular pathways, and varying with the disease context. An understanding of polyglutamine disease mechanisms is critical for development of novel therapeutics. Here we summarise theories of molecular pathogenesis, and examine ways in which this knowledge is being harnessed for therapy, with reference to work under way at the University of Cape Town. Despite a plethora of preclinical data, clinical trials of therapies for polyglutamine diseases have had only limited success. However, recently initiated trials, including those using gene silencing approaches, should provide valuable insights into the safety and efficacy of therapiesdirectly targeting polyglutamine pathogenesis. This is particularly relevant in the South African context, where the frequencies of 2 polyglutamine diseases, spinocerebellar ataxia types 1 and 7, are among the highest globally.
Polyglutamine (polyQ) diseases are inherited neurodegenerative conditions caused by expansion of a coding trinucleotide CAG repeat, which is translated into an abnormally long polyQ tract in the mutant protein.1 There are currently 9 known polyQ diseases: Huntington disease (HD), dentatorubral-pallidoluysian atrophy (DRPLA), spinal bulbar muscular atrophy (SBMA) and 6 spinocerebellar ataxias (SCA 1, 2, 3, 6, 7 and 17). The molecular diagnostics laboratory of the National Health Laboratory Service in Cape Town is the only centre in South Africa offering diagnostic testing for the polyQ SCAs, and works in close collaboration with the Division of Human Genetics at the University of Cape Town (UCT) to provide patients with the opportunity to participate in ground-breaking research into pathogenesis and treatment.
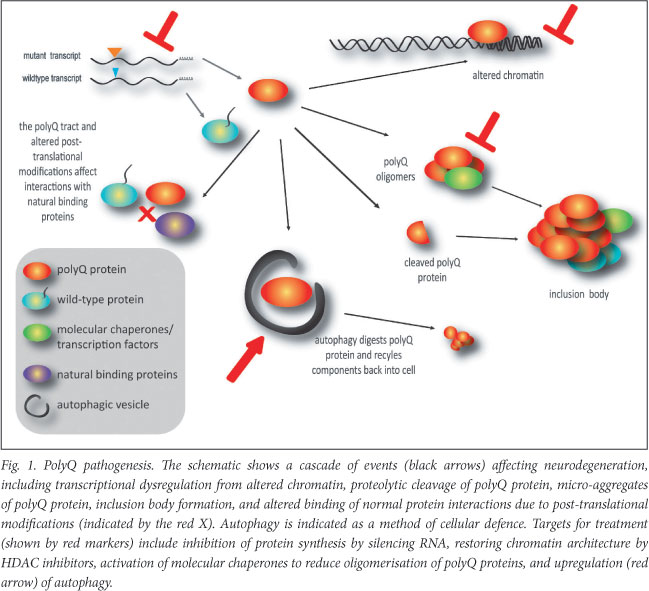
Despite genetic and clinical heterogeneity, the presence of a common mutation suggested that a common pathogenic mechanism must link all 9 diseases. However, emerging evidence indicates that polyQ pathogenesis is more complex (Fig. 1). Here we summarise theories of pathogenesis and discuss how this knowledge underpins the development of novel therapeutic strategies.
PolyQ disease pathogenesis mechanisms
Aggregation and impairment of proteasome clearance
Early studies of polyQ disease revealed large nuclear inclusion bodies containing proteolytically cleaved mutant protein as a common pathogenic marker. These aggregates recruit nuclear proteins, in addition to proteins containing polyQ tracts. They are marked for ubiquitin proteasome degradation but for unknown reasons are not degraded.This suggests that the structure of the aggregates renders them resistant to the misfolded protein clearance pathway, leading to cellular dysfunction.2 This theory could account for the susceptibility of neurons associated with all 9 disorders, since these highly metabolically active cells may be more sensitive to perturbations in cellular machinery.
Further investigations - including the observation that their formation does not entirely correlate with neuronal susceptibility - have contradicted the theory of inclusion body toxicity, with increasing evidence suggesting that they might instead serve a protective role within cells. It is now generally accepted that the toxic species are smaller, less visible micro-aggregates. Nevertheless, inclusions remain a hallmark of polyQ disease pathogenesis.
Autophagy
Impairment of autophagy, the process by which the cell recycles intracellular components, has been implicated in polyQ disorders.3 Although the mechanism is unclear, upregulation of autophagy alleviates cytotoxicity in models of polyQ disease. This provides a common theory of polyQ pathogenesis since loss of autophagy causes general neurodegeneration in mice.4 However, all studies to date have been performed in models over-expressing the mutant protein, and therefore studies in more representative models of polyQ disease are crucial.
Protein-protein interactions/post-translational mechanisms
The expanded polyQ tract imparts a conformational change to the mutant protein and therefore directly affects interactions with its native binding partners. In addition, post-translational modifications such as phosphorylation, SUMOylation and acetylation of specific residues in several polyQ proteins can be altered in the presence of the mutation, affecting interactions with their natural binding proteins. Importantly, direct inhibition of these post-translational alterations reduces mutant protein toxicity (reviewed by La Spada and Taylor5).
Transcriptional alterations
Many polyQ proteins function in transcriptional regulation,6 and several reports have identified transcriptional changes in molecular chaperones and genes involved in neuronal differentiation, often observed prior to cellular dysfunction. However, studies have not revealed a common subset of dysregulated transcripts. How then do the polyQ proteins confer transcriptional alterations if not through the shared mutation? A possible answer is that transcriptional effects depend on the cellular context of the mutant polyQ protein. Each native protein has distinct regulatory mechanisms in different neuronal sub-types; thus, the mutation is likely to lead to transcriptional dysregulation in the neuronal subset most susceptible to changes in that protein. One mechanism for this change may be chromatin modification. If chromatin architecture is unique to each neuronal subset they may be differentially susceptible to mutations in each polyQ protein.6
Loss of function and additional pathogenic mechanisms
Although polyQ diseases are caused by a toxic gain-of-function of the mutant protein, it is likely that loss of function also contributes to pathogenesis, since knockout models of polyQ disease show neurodegeneration and polyQ inclusions recruit the native protein, impairing its normal function. Thus, understanding normal protein function is critical in understanding disease pathogenesis. A number of additional disease mechanisms have been proposed including mitochondrial dysfunction, enhanced apoptosis and excitotoxicity caused by glutamate receptor overactivation.
Given the multitude of cellular insults triggered by expanded polyQ tracts, developing an effective therapy appears a daunting prospect. However, based on current theories of pathogenesis, several proof-of-principle treatments have already been demonstrated.
Therapeutic strategies
Historically, treatment for polyQ diseases has focused on symptomatic management, rather than disease modification. Understanding the molecular mechanisms of pathogenesis offers the opportunity to prevent or delay disease symptom onset, through the identification of targets within the disease pathway which may be amenable to therapy (Fig. 1). Experimental therapies fall into 2 categories - those aimed at removing the burden of accumulated mutant protein and those targeting downstream effects of the mutant protein.
Therapies targeting polyglutamine proteins Gene silencing
Given the complexities of polyQ pathogenesis, one logical approach is to suppress production of the mutant protein upstream of its deleterious effects. Further, silencing the mutant gene, while retaining wild-type expression may be of therapeutic benefit. This has led to the development of gene silencing therapies, using either RNA interference (RNAi) or antisense oligonucleotide (AON) approaches. Both have shown promise in studies in animal models of polyQ diseases, including SBMA, SCA1, and HD - rescuing toxicity, preventing apoptosis, and allowing for the development of sophisticated delivery mechanisms.7 Researchers at UCT, in collaboration with the Universities of Oxford and the Witwatersrand, were the first to demonstrate the potential for RNAi-based gene silencing in the South African SCA7 population. However, a number of challenges must be addressed before these results can be translated to the clinic.
Evidence from knockout animal models suggests that several polyQ genes are critical in development and central nervous system (CNS) function and therefore that allele-specific therapies may be required. This approach, which relies on the presence of targetable sequence differences in order to discriminate between the normal and mutated transcripts, is technically challenging, and likely to limit eligibility for therapies, since it relies on single nucleotide polymorphisms (SNPs) in linkage disequilibrium with the polyQ expansion, or differences in CAG repeat length to accomplish sequence discrimination. Two recent studies, encouraging efforts to continue to develop gene silencing therapies for the polyQ diseases, indicate that non-allele-specific approaches are better tolerated in HD and SCA3 mouse models than was originally thought;8,9 and that allele-specific silencing may provide therapy in a majority of European HD patients using relatively few disease-linked SNPs.10
Clearing misfolded mutant protein and inhibiting aggregate formation
Decreasing mutant protein levels may also be achieved by enhancing polyQ protein clearance, through upregulation of the two main cellular pathways responsible for degradation of misfolded proteins - the ubiquitin-proteasome system (UPS) and autophagy. While impairment of the UPS has been observed in several polyQ diseases, upregulation of this pathway is technically challenging and attention has therefore shifted to enhancing autophagy. Activation of autophagy using the mTOR inhibitor rapamycin has been shown to reduce mutant protein toxicity in models of HD and SCA3, suggesting that small molecules capable of inducing the autophagic cascade may be of therapeutic benefit.11,12
Another approach to clear mutant polyQ proteins is via molecular chaperones capable of promoting refolding of misfolded proteins. Overexpression of heat shock proteins Hsp27 and Hsp104 suppresses neurotoxicity in HD models.13 However, global induction of the heat shock response (HSR) is of only transient benefit, suggesting that impairment of the HSR may occur during polyQ disease progression.14 These findings motivate for the development of combinatorial therapies such as histone deacetylase (HDAC) inhibitors capable of enhancing HSR gene transcription.
Direct methods of preventing polyQ protein aggregation using molecules such as Congo Red and cystamine, which act to stabilise the native non-toxic conformation of the protein, prevent cross-linking of the mutant isoform, or promote clearance from the cell, have also been attempted. All have demonstrated some therapeutic benefit but efficacy and delivery remain to be optimised.15
Therapies targeting downstream effects of polyQ proteins
Transcriptional dysregulation
The inhibition of HDACs, repressors of transcription, has been proposed as a method for mitigating transcriptional dysregulation. A variety of small-molecule HDAC inhibitors have been tested as anti-cancer drugs and several have been shown to rescue polyQ disease phenotypes.16 In particular, the discovery that HD mice, engineered to be deficient in HDAC4, show a phenotypic improvement suggests that this may be a promising therapeutic approach.
Upregulation of specific genes known to have a neuroprotective role (e.g. brain derived neurotrophic factor) has also shown beneficial effects. However, this requires a thorough understanding of the functions of downstream targets of transcriptional regulation by polyQ proteins, in order to identify the most promising therapeutic targets.
Additional therapeutic approaches
A number of additional therapeutic approaches have been proposed, which aim to reverse the downstream cellular effects of mutant polyQ proteins. These include compounds capable of alleviating energy metabolism defects or mitigating oxidative stress. Riluzole and remacemide, which mitigate excitotoxicity, have achieved some success in HD mice, although these results have not been successfully reproduced in clinical trials.17 Anti-apoptotic drugs such as minocycline and caspase inhibitors have also shown promise in polyQ animal models, although data from long-term studies will be required in order to rule out possible side-effects associated with suppression of these key cellular pathways.
Clinical considerations
Despite the plethora of preclinical investigations, few have progressed to clinical trials. The low incidence, late onset, slow progression and clinical heterogeneity of polyQ diseases make demonstration of clinical efficacy particularly difficult.
A number of major technical issues remain to be addressed, including questions of dosage and delivery - particularly pertinent in CNS conditions, which require delivery across the blood-brain barrier, and rapid attenuation in the event of adverse effects. There are also more challenging tasks - designing therapies which treat the maximum number of patients, without compromising on off-target effects, immunogenicity or allele specificity (in the case of gene silencing); and balancing the benefits of relatively broad-spectrum therapeutics (such as HSR induction or RNAi) with their potential to disrupt normal cellular processes.
Nonetheless, the recent progress of RNAi to the clinic suggests that effective therapies for polyQ diseases may be within reach. At the time of writing, 14 clinical trials involving RNAi are ongoing, although none yet involve the CNS.18 AONs are also being tested in trials for Duchenne muscular dystrophy and cancer (http:// www.clinicaltrials.gov), and the first clinical study of AONs in neurodegenerative disease has been initiated.19
Several therapies designed to target additional polyQ pathogenic mechanisms, including mitigators of exicitotoxicity, UPS activators, anti-apoptotics, and modulators of transcriptional activity, are undergoing clinical trials (http://www.clinicaltrials.gov). SBMA, unique among the polyQ diseases by virtue of its X-linked mode of inheritance, is also the target of novel therapeutic strategies involving androgen deprivation, but with limited efficacy to date.
Results from these trials should provide valuable insight into how to safely and effectively deliver therapies to the human CNS. However, critical to success will be the generation of more accurate in vitro and in vivo models of disease (see below).
The South African context
The development of effective therapies for polyQ diseases is particularly relevant to South Africa, where the frequencies of SCA1 and SCA7 are among the highest globally.20 Researchers at UCT have previously reported founder effects in the mixed-ancestry population (SCA121 and HD22) and the indigenous black African population (SCA723), with further research ongoing into the HD founder haplotypes. This phenomenon offers the ideal genetic background for the development of allele-specific gene-silencing-based therapies in South Africa, allowing patients to be treated by targeting a single disease-linked SNP.24
The neurodegenerative disorders research team at the UCT Division of Human Genetics, in collaboration the University of Oxford, has recently established a ground-breaking new disease model for SCA7 using patient-derived cells to generate induced pluripotent stem cells (iPSCs). This offers the first human disease cell model to test and refine the novel therapies developed in the UCT laboratory.
Conclusion
The identification of a common pathogenic mutation underlying the clinically and genetically heterogeneous polyQ diseases suggested initially that common therapies could be easily designed. The reality is vastly different. As the molecular basis of polyQ diseases is revealed in ever-increasing complexity, it is clear that the task of developing therapies is not straightforward. Nevertheless, the identification of numerous therapeutic targets and development of a human cell-based model of disease, offers the potential to develop effective treatments for polyQ diseases.
Acknowledgements
LW is funded by the National Research Foundation (NRF), the Harry Crossley Foundation and the Commonwealth Scholarship Commission UK. At the time of writing, JS was an Oxford Nuffield Medical Fellow. Work in the UCT laboratory is funded by the UCT Faculty of Health Sciences Research Council and the NRF. Work in the MJAW Laboratory is funded by the UK Medical Research Council, The Wellcome Trust, Parkinson's UK, Ataxia UK, and the John Fell Fund. The financial assistance of the NRF of South Africa towards this research is hereby acknowledged. Opinions expressed, and conclusions arrived at, are those of the authors and are not necessarily to be attributed to the NRF.
References
1. Durr A. Autosomal dominant cerebellar ataxias: polyglutamine expansions and beyond. Lancet Neurol 2010;9:885-894. [http://dx.doi.org/10.1016/S1474-4422(10)70183-6] [ Links ]
2. Bence NF, Sampat RM, Kopito RR. Impairment of the ubiquitin-proteasome system by protein aggregation. Science 2001;292:1552-1555. [http://dx.doi.org/10.1126/science.292.5521.1552] [ Links ]
3. Ravikumar B, Vacher C, Berger Z, et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet 2004;36:585-595. [http://dx.doi.org/10.1038/ng1362] [ Links ]
4. Rubinsztein DC. The roles of intracellular protein-degradation pathways in neurodegeneration. Nature 2006;443:780-786. [http://dx.doi.org/10.1038/nature05291] [ Links ]
5. La Spada AR, Taylor JP. Repeat expansion disease: progress and puzzles in disease pathogenesis. Nat Rev Genet 2010;11:247-258.[http://dx.doi.org/10.1038/nrg2748] [ Links ]
6. Helmlinger D, Tora L, Devys D. Transcriptional alterations and chromatin remodeling in polyglutamine diseases. Trends Genet 2006;22:562-570. [http://dx.doi.org/10.1016/j.tig.2006.07.010] [ Links ]
7. Scholefield J, Wood MJA. Therapeutic gene silencing strategies for polyglutamine disorders. Trends Genet 2010;26:29-38. [http://dx.doi.org/10.1016/j.tig.2009.11.005] [ Links ]
8. Alves S, Nascimento-Ferreira I, Dufour N, et al. Silencing ataxin-3 mitigates degeneration in a rat model of Machado-Joseph disease: no role for wild-type ataxin-3? Hum Mol Genet 2010;19:2380-2394. [http://dx.doi.org/10.1093/hmg/ddq111] [ Links ]
9. Boudreau RL, McBride JL, Martins I, et al. Nonallele-specific silencing of mutant and wild-type huntingtin demonstrates therapeutic efficacy in Huntington's disease mice. Mol Ther 2009;17:1053-1063. [http://dx.doi.org/10.1038/mt.2009.17] [ Links ]
10. Pfister EL, Kennington L, Straubhaar J, et al. Five siRNAs targeting three SNPs may provide therapy for three-quarters of Huntington's disease patients. Curr Biol 2009;19:774-778. [http://dx.doi.org/10.1016/j.cub.2009.03.030] [ Links ]
11. Renna M, Jimenez-Sanchez M, Sarkar S, Rubinsztein DC. Chemical inducers of autophagy that enhance the clearance of mutant proteins in neurodegenerative diseases. J Biol Chem 2010;285:11061-11067. [http://dx.doi.org/10.1074/jbc.R109.072181] [ Links ]
12. Menzies FM, Rubinsztein DC. Broadening the therapeutic scope for rapamycin treatment. Autophagy2010;6:286-287. [http://dx.doi.org/10.4161/auto.6.2.11078] [ Links ]
13. Perrin V, Regulier E, Abbas-Terki T, et al. Neuroprotection by Hsp104 and Hsp27 in lentiviral-based rat models of Huntington's disease. Mol Ther 2007;15:903-911. [http://dx.doi.org/10.1038/mt.sj.6300141] [ Links ]
14. Labbadia J, Cunliffe H, Weiss A, et al. Altered chromatin architecture underlies progressive impairment of the heat shock response in mouse models of Huntington disease. J Clin Invest 2011;121:3306 3319.[http://dx.doi.org/10.1172/JCI57413] [ Links ]
15. Bauer PO, Nukina N. The pathogenic mechanisms of polyglutamine diseases and current therapeutic strategies. J Neurochem 2009;110:1737-1765. [http://dx.doi.org/10.1111/j.1471-4159.2009.06302.x] [ Links ]
16. Ross CA, Tabrizi SJ. Huntington's disease: from molecular pathogenesis to clinical treatment. Lancet Neurol 2011;10:83-98. [http://dx.doi.org/10.1016/S1474-4422(10)70245-3] [ Links ]
17. Landwehrmeyer GB, Dubois B, de Yebenes JG, et al. Riluzole in Huntington's disease: a 3-year, randomized controlled study. Ann Neurol 2007;62:262-272. [http://dx.doi.org/10.1002/ana.21181] [ Links ]
18. Vaishnaw AK, Gollob J, Gamba-Vitalo C, et al. A status report on RNAi therapeutics. Silence 2010;1:1 13. [http://dx.doi.org/10.1186/1758-907X-1-14] [ Links ]
19. Sah DWY, Aronin N. Oligonucleotide therapeutic approaches for Huntington disease. J Clin Invest 2011;121:500-507. [http://dx.doi.org/10.1172/JCI45130] [ Links ]
20. Bryer A, Krause A, Bill P, et al. The hereditary adult-onset ataxias in South Africa. J Neurol Sci 2003;216:47-54. [http://dx.doi.org/10.1016/S0022-510X(03)00209-0] [ Links ]
21. Ramesar RS, Bardien S, Beighton P, Bryer A. Expanded CAG repeats in spinocerebellar ataxia (SCA1) segregate with distinct haplotypes in South African families. Hum Genet 1997;100:131-137. [http://dx.doi.org/10.1007/s004390050478] [ Links ]
22. Scholefield J, Greenberg J. A common SNP haplotype provides molecular proof of a founder effect of Huntington disease linking two South African populations. Eur J Hum Genet 2007;15:590-595. [http://dx.doi.org/10.1038/sj.ejhg.5201796] [ Links ]
23. Greenberg J, Solomon G, Vorster A, Heckmann J, Bryer A. Origin of the SCA7 gene mutation in South Africa: implications for molecular diagnostics. Clin Genet 2006;70:415-417. [http://dx.doi.org/10.1111/j.1399-0004.2006.00680.x] [ Links ]
24. Scholefield J, Greenberg LJ, Weinberg MS, Arbuthnot PB, Abdelgany A, Wood MJA. Design of RNAi hairpins for mutation-specific silencing of ataxin-7 and correction of a SCA7 phenotype. PLoS ONE 2009;4:e7232. [http://dx.doi.org/10.1371/journal.pone.0007232] [ Links ]
Accepted 26 February 2012.
Corresponding author: M J A Wood (matthew.wood@dpag.ox.ac.uk)


{kind=link}