










Serviços Personalizados
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkSAMJ: South African Medical Journal
versão On-line ISSN 2078-5135
versão impressa ISSN 0256-9574
SAMJ, S. Afr. med. j. vol.102 no.6 Pretoria Jun. 2012
FORUM
REVIEW
Haematological complications of HIV infection
Jessica Opie
Senior haematology pathologist in the National Health Laboratory Service and Division of Haematology, Groote Schuur Hospital and University of Cape Town
ABSTRACT
South Africa is in the midst of the world’s largest human immune deficiency virus (HIV) epidemic with an estimated 5.6 million people infected. Haematological manifestations of HIV are common and diverse, occurring at all stages of infection. Haematological emergencies occurring in this setting include the high-grade lymphomas, particularly Burkitt lymphoma, and thrombotic thrombocytopenic purpura (TTP). Immune thrombocytopenic purpura (ITP), opportunistic infections and drug side-effects are also frequent causes of cytopenias. A bone marrow biopsy has a high diagnostic utility in HIV patients presenting with unexplained cytopenias and/or fevers. It is not widely realised that HIV is also a prothrombotic state with an increased incidence of thromboembolic disease. Highly active antiretroviral therapy (HAART) is now widely available in South Africa and is a crucial adjunct to therapy of haematological complications. Medical professionals across all disciplines need to be alert to the haematological complications of HIV infection.
More people live with HIV in South Africa (SA) than any other country in the world. In 2010 approximately 68% of all people infected with HIV resided in sub-Saharan Africa, which has only 12% of the world's population. The roll-out of antiretroviral therapy (ART) commenced in the public sector in SA in 2004 and has increased dramatically since then. According to UNAIDS World AIDS Day Report 2011, 40 - 59% of eligible South Africans were receiving ART by the end of 2010, which is the largest comprehensive ART programme in the world.1 Although widespread access to ART has improved quality of life and reduced AIDS-related deaths and HIV transmission, the challenges to the healthcare sector remain considerable.
Cytopenias
Cytopenias are one of the most common complications of HIV and may be broadly classified as being due either to a bone marrow production defect or to increased peripheral loss or destruction of blood cells. Anaemia is the most common cytopenia and occurs in up to 95% of HIV patients during their disease course.2 There are a multitude of possible causes of anaemia in HIV (Table 1).
The main mechanism for anaemia of chronic disease is perturbed bone marrow cytokine homeostasis. HIV is cytotoxic to T-helper lymphocytes, which in turn leads to dysregulation of B cells and altered release of cytokines. HIV-infected T cells directly suppress growth of bone marrow progenitors, thus suppressing haemopoiesis. CD4, the cell-surface receptor target of HIV, is carried by T-helper lymphocytes, monocytes and microvascular endothelial cells which are prevalent in marrow. The infection of monocytes in the marrow further alters release of cytokines, which indirectly suppress the capacity for haemopoietic progenitor cells to adequately respond to anaemia and other peripheral cytopenias. This explains why in most patients with advanced HIV pancytopenia is the rule.3
Cytopenias due to drug therapy (Table 2) add to the diagnostic challenge in HIV patients with cytopenias and may require drug cessation. Culprits include zidovudine (AZT) and stavudine (d4T), co-trimoxazole (Bactrim) and the antituberculosis drugs isoniazid, rifampicin and rifabutin.4 In patients presenting with anaemia, baseline haematinic studies including serum ferritin, serum B12 and red-cell folate are essential, and significant deficiencies should routinely be excluded. Although iron deficiency typically causes a microcytic anaemia and B12 and folate deficiency a macrocytic anaemia, this distinction often becomes blurred in HIV patients who typically become macrocytic while receiving ART. Highly active ART (HAART) is an important intervention in cytopenic HIV patients, as it alleviates the cytokine disturbances. However, if patients develop a new cytopenia while taking HAART, then a drug-induced cytopenia must be considered and a change of HAART regimen may be required.
Infections
Opportunistic infections such as Mycobacterium tuberculosis, M. avium complex (MAC) and Cryptococcus neoformans can cause cytopenias by infiltrating the bone marrow and causing reactive granuloma formation (Fig. 1). A bone marrow biopsy can provide a morphological diagnosis within a few days. A study by the University of Cape Town (UCT) haematology department at Groote Schuur Hospital (GSH), which included 147 patients, showed the high diagnostic utility of a bone marrow biopsy in HIV patients presenting with unexplained cytopenias and/or fever.5 A bone marrow biopsy gave an overall diagnostic yield of 47% with a unique diagnosis, defined as a diagnosis not made by any other modality and which resulted in a change in patient management, made in 33% of patients. Mycolytic cultures of the blood and bone marrow were important adjuncts which added to the high diagnostic yield of tuberculosis (TB) in this study. However, cultures have a much longer turnaround time than morphology. Notably, patients in the study group generally had advanced HIV disease (the median CD4 count was 73 x 106/l; with 43% of cases having CD4 < 50 x 106/l), which probably contributed towards the high diagnostic yield. The most common unique diagnoses made were disseminated mycobacterial infection and immune thrombocytopenic purpura (ITP) (14% each), followed by malignancies (4%). The unexpected malignancies found on bone marrow examination included 4 cases of primary bone marrow Hodgkin lymphoma (HL) and 1 case of non-Hodgkin lymphoma (NHL). In the marrows that were not specific diagnostically, morphological dysplasia of one or more cell lines was a frequent finding. HIV dysplasia, which is a well-recognised entity, is non-clonal and must be distinguished from the preleukaemic myelodysplastic syndrome, although the morphological features are often indistinguishable. Morphological features of HIV dysplasia become more marked as the disease progresses.
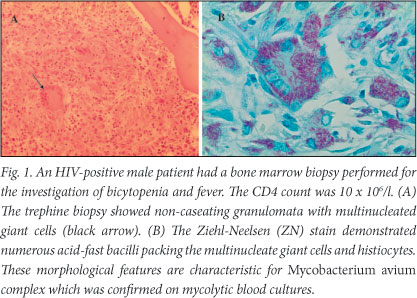
Immunosuppressed HIV patients are predisposed to parvovirus B19 infection, which should be suspected in all cases of isolated severe anaemia, usually with preserved platelet and white cell counts. Parvovirus B19 is a DNA virus which is usually acquired through the respiratory tract, and infects red-cell precursors in the bone marrow. The virus then replicates and lyses these red-cell precursors, leading to a marked reduction in erythroid activity in the marrow. There is an absent or very low reticulocyte response. There are characteristic morphological features on the bone marrow biopsy such as red-cell maturation arrest and giant pronormoblasts, but the bone marrow biopsy is not required for diagnosis. Polymerase chain reaction (PCR) is the diagnostic test of choice. Serology is often unhelpful because during advanced HIV infection, patients are unable to mount a serological response. Management includes HAART, red-cell transfusions and intravenous immunoglobulin therapy, if available.6
Immune thrombocytopenic purpura
ITP occurs in up to 30% of HIV patients and is the most common cause of thrombocytopenia in HIV. Although the clinical presentation is similar to non-HIV-associated ITP, the mechanism is unique and thought to be due to an HIV-induced auto-antibody generated against an amino-acid sequence within the platelet surface glycoprotein IIIa.2 Many patients present with ITP as the first manifestation of HIV, although it can occur in both early and advanced disease. Management includes HAART and prednisone at 1 mg/kg/day if the platelet count is <30 x 109/l. If the platelet count does not improve after 2 weeks, referral to a tertiary haematology centre is advised. For patients unresponsive to initial doses of prednisone, the further management at our centre is to double the prednisone dose and if there is still no response, splenectomy is then considered.
In our recent HIV bone marrow study, ITP was a frequent diagnosis, but the bone marrow findings did not add any further diagnostic information.5 This supports the argument that where the history, clinical examination and laboratory findings are compatible with the diagnosis of ITP, a bone marrow biopsy is unnecessary.
Thrombotic thrombocytopenic purpura (TTP)
TTP is a potentially lethal condition which occurs when ultra-large Von Willebrand factor (VWF) multimers cause platelet microthrombi in arterioles and capillaries. These platelet microthrombi lead to intravascular haemolysis and organ ischaemia. The red cells are mechanically sheared as they pass through the platelet thrombi in the microcirculation, leading to the characteristic red-cell fragments or 'schistocytes' on the blood smear (Fig. 2). HIV is the most common virus precipitating TTP. Other precipitants include pregnancy, autoimmune disorders, malignancies and drugs, e.g. clopidogrel and statins.7 TTP is characterised by thrombocytopenia, micro-angiopathic haemolytic anaemia (MAHA), fluctuating neurological signs, fever and renal impairment. In practice, however, not all features need to be present. British guidelines on the diagnosis and management of TTP8 state that severe thrombocytopenia plus significant red-cell fragmentation on the blood smear, and where other causes of MAHA, such as malignant hypertension and disseminated intravascular coagulation (DIC), have been excluded, is compatible with the diagnosis of TTP. Coagulation parameters are expected to be normal, in contrast to DIC, and the lactate dehydrogenase (LDH) is markedly elevated. Renal function should routinely be assessed and may be deranged; however, in acute severe renal impairment, an alternative diagnosis such as haemolytic uraemic syndrome should be considered. HIV-associated TTP typically occurs in young African females not on HAART and with high viral loads.9
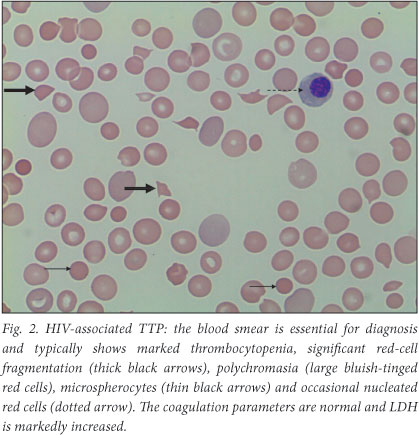
In First World settings, the standard of care for HIV-associated TTP is plasma exchange in conjunction with steroids and HAART; however, plasmapharesis is not widely available in resource-poor settings. Our department showed that HIV-associated TTP is highly responsive to plasma infusion therapy;10 daily fresh-frozen plasma (FFP) infusions at 30 ml/kg/day in conjunction with prednisone 1 mg/kg/day resulted in excellent response rates (95%). HAART should be simultaneously (re-)initiated.9 This protocol is feasible in cost-limited settings and has become the standard of care for HIV-associated TTP in SA. FFP can be commenced at a local hospital, but all TTP patients should be referred to a tertiary centre for definitive management. The small proportion of TTP cases resistant to FFP then require plasmapharesis; other aetiologies such as underlying malignancy and autoimmune diseases should then be explored.
Lymphoma
HIV is associated with a markedly increased risk of malignancies, particularly high-grade B-cell lymphoma (Table 3). HIV infection increases NHL indicidence by 60 - 200-fold. The postulated mechanisms include chronic antigen stimulation, cytokine deregulation and Epstein-Barr virus (EBV) and human herpes virus 8 (HHV8) co-infection. EBV has been identified in up to 40% of HIV-related lymphomas.11

Since the widespread introduction of HAART in SA in 2004, there has been an increase in HIV patients presenting at GSH with lymphoma with a particularly rapid increase in the incidence of Burkitt lymphoma/leukaemia (BL) (Professor N Novitzky, 2012 unpublished observation) and diffuse large B-cell lymphoma.12
BL is the most aggressive tumour known. In Burkitt lymphoma, the disease is localised to lymph nodes, while in Burkitt leukaemia, it is disseminated in the blood and bone marrow. BL is characterised by translocations of the c-MYC gene on chromosome 8 which lead to uncontrolled cell proliferation. Cytogenetic or fluorescent in situ hybridisation (FISH) analysis, available at tertiary centres in South Africa, usually reveals the characteristic translocation t(8;14).
In HIV patients high-grade NHL typically presents with advanced clinical stage, bulky disease and a markedly elevated LDH. Many BL patients also present with, or develop, potentially lethal tumour lysis syndrome because of the very high cell turnover with associated simultaneous rapid cellular breakdown. Tumour lysis syndrome is characterised by hyperkalaemia, hyperuricaemia, hyperphosphataemia and renal dysfunction, and requires meticulous management including vigorous hydration, careful fluid balance, urinary alkalinisation and allopurinol. As a general principle, all HIV patients with lymphoma should receive HAART in conjunction with chemotherapy.
Treatment for BL requires very intensive combination chemotherapy regimens with higher doses than those for acute leukaemias and other high-grade lymphomas. Because of the high incidence of BL patients at GSH, our bed capacity has been overwhelmed. A cost-saving and bed-saving therapeutic strategy has thus been developed to allow BL patients to be effectively treated on a short, mainly outpatient protocol. This ultrashort dose-dense regimen ('Cape Town regimen') has shown considerable cost saving compared with standard protocols and comparable disease-free survival and treatment-related mortality,13 and may become a model for therapy in other centres confronting similar challenges.
Another HIV-associated NHL occurring with increasing frequency at GSH is plasmablastic lymphoma (PL). This is an aggressive disease originally described in the oral cavity and associated with low CD4 counts of <100 x 106/l. According to the World Health Organization (WHO), PL is rare.11 However, PL is not rare in our setting. A recent 4-year review at the GSH oncology department included 28 patients with PL; 25 patients were HIV-positive and 3 patients HIV-negative. The most common sites of involvement were the oral cavity, maxillary sinus and nasopharynx (32%) followed by bone (28%) and skin and subcutaneous lesions (20%). Although PL generally has a poor outcome, it responds to chemotherapy and radiotherapy with 40% of patients in this study surviving for the 9-month follow-up period.14
Other HHV8-associated malignancies occurring with increased frequency in HIV infection include large B-cell lymphoma arising in HHV8-associated multicentric Castleman disease, Kaposi sarcoma and primary effusion lymphoma.11 Primary effusion lymphoma is exceedingly rare in our centre with only one possible case in the last 10 years (Dr Andrew McDonald, personal communication). However NHL arising in Castleman disease is not infrequent and classically presents with enlarging lymph nodes and massive splenomegaly. The disease responds well to standard cyclophosphamide, doxorubicin, vincristine, prednisolone ('CHOP') chemotherapy.
HIV is known to be associated with a 5 - 10-fold increased incidence of HL, and in these patients there is a higher incidence of bone marrow involvement compared with HIV-negative patients (Fig. 3) (Table 3). HIV patients also have an increased incidence of primary bone marrow HL.15 Primary bone marrow HL, where there is no lymphadenopathy or organ involvement to suggest lymphoma, generally presents with cytopenias and 'B symptoms' (such as weight loss and night sweats) and has an aggressive clinical course.
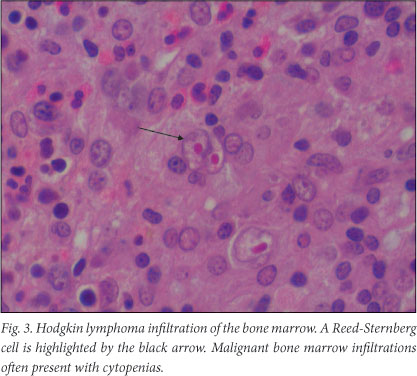
An adequate tissue biopsy sample is imperative for accurate histopathological diagnosis in lymphoma, and when the NHL is disseminated, blood/body fluid/bone marrow aspirate samples can be submitted for immunophenotyping by flow cytometric analysis, which greatly assists diagnosis.
Thrombosis
HIV is a prothrombotic condition associated with a 2 - 10-fold increased incidence of venous thromboembolism (VTE) compared with the HIV-negative population of the same age.16 The risk is highest with advanced disease and co-existing infections and malignancies. There is disruption of the normal balance of coagulation factors with an increase in prothrombotic proteins such as VWF and reduced natural anticoagulant proteins such as protein S and protein C. While most abnormal coagulation factors/markers improve after starting HAART, the disturbances fail to normalise completely.17 HIV patients also have a higher incidence of the lupus anticoagulant and antiphospholipid antibodies than the general population, which may contribute to a hypercoagulable state. Therefore, clinicians should have a high index of suspicion for VTE in HIV patients and prophylactic anticoagulation should be considered for inpatients.
Conclusions
Haematological abnormalities in HIV are frequent and varied, with a wide clinical spectrum of disease. Usually multiple factors contribute to the haematological derangements, and a meticulous, logical approach, including consideration of all possible contributors such as haematinic deficiencies, drug effects and bone marrow infiltration, should be considered. Institution of HAART is a general rule of thumb and clinical management needs to be tailored to the particular complication. Life-threatening medical emergencies include TTP and tumour lysis syndrome in high-grade lymphoma; these require urgent hospitalisation and management.
References
1. Joint United Nations Programme on HIV/AIDS. UNAIDS World AIDS Day Report 2011. Geneva. http://www.unaids.org/ (accessed 5 January 2012). [ Links ]
2. Baker K. The hematologic complications of HIV infection. ASH Education Program 2003;1:299. [ Links ]
3. Moses A, Nelson J, Bagby GC. The influence of human immunodeficiency virus-1 on hematopoiesis. Blood 1998;91(5):1479-1495. [ Links ]
4. Division of Pharmacology, Faculty of Health Sciences, University of Cape Town. General anti-infectives for systemic use. South African Medicines Formulary. Cape Town: HMPG, 2008:264. [ Links ]
5. Van Schalkwyk WA, Opie J, Novitzky N. The diagnostic utility of bone marrow biopsies performed for the investigation of fever and/or cytopenias in HIV-infected adults at Groote Schuur Hospital, Western Cape, South Africa. Int J Lab Hematol 2011;33(3):258-266. [http://dx.doi.org/10.1111%2Fj.1751-553X.2010.01280.X] [ Links ]
6. Sawada K, Fujishima N, Hirokawa M. Acquired pure red cell aplasia: updated review of treatment. Br J Haematol 2008;142(4):505-514. [http://dx.doi.org/10.1111%2Fj.1365-2141.2008.07216.x] [ Links ]
7. Hoffbrand A, Catovsky D, Tuddenham E. Postgraduate Haematology. 5th ed. Oxford: Blackwell Publishing, 2005. [http://dx.doi.org/10.1002%2F9780470987056] [ Links ]
8. Allford SL, Hunt BJ, Rose P, Machin SJ, Haemostasis and Thrombosis Task Force, British Committee for Standards in Haematology. Guidelines on the diagnosis and management of the thrombotic microangiopathic haemolytic anaemias. Br J Haematol 2003;120(4):556 573.[http://dx.doi.org/10.1046%2Fj.1365-2141.2003.04049.x] [ Links ]
9. Hart D, Sayer R, Miller R, et al. Human immunodeficiency virus associated thrombotic thrombocytopenic purpura - favourable outcome with plasma exchange and prompt initiation of highly active antiretroviral therapy. Br J Haematol 2011;153(4):515-519. [http://dx.doi.org/10.1111/j.1365-2141.2011.08636.x] [ Links ]
10. Novitzky N, Thomson J, Abrahams L, du Toit C, McDonald A. Thrombotic thrombocytopenic purpura in patients with retroviral infection is highly responsive to plasma infusion therapy. Br J Haematol 2005;128(3):373-379.[http://dx.doi.org/10.1111%2Fj.1365-2141.2004.05325.x] [ Links ]
11. World Health Organization Classification of Tumours of Haemopoietic and Lymphoid Tissue. 4th ed. Lyon, France: International Agency for Research on Cancer, 2008. [ Links ]
12. Mohammed Z, Novitzky N, McDonald A. Audit of diffuse large B-cell lymphoma in HIV-positive patients at Groote Schuur Hospital. 3rd South African Haematology-Oncology Symposium, Johannesburg, 2009. [ Links ]
13. Gerdener T, Novitzky N, McDonald A, du Toit C. Evaluation of an ultra-short, dose-dense regimen for the treatment of Burkitt lymphoma in adults and adolescents: the Cape Town experience. 5th South African Haematology-Oncology Symposium. Johannesburg, 2011 (Abstract: 30). [ Links ]
14. Mohammed Z, Novitzky N, McDonald A. Plasmablastic lymphoma: a single institution experience. 3rd South African Haematology-Oncology Symposium, Johannesburg, 2009. [ Links ]
15. Shah BK, Subramaniam S, Peace D, Garcia C. HIV-associated primary bone marrow Hodgkin's lymphoma: a distinct entity? J Clin Oncol 2010;28(27):e459-60. [http://dx.doi.org/10.1200%2FJCO.2010.28.3077] [ Links ]
16. Bibas M, Biava G, Antinori A. HIV-associated venous thromboembolism. Mediterr J Hematol Infect Dis 2011;3(1):e2011030. [http://dx.doi.org/10.4084%2Fmjhid.2011.030] [ Links ]
17. Jong E, Louw S, van Gorp EC, Meijers JC, ten Cate H, Jacobson BF. The effect of initiating combined antiretroviral therapy on endothelial cell activation and coagulation markers in South African HIV-infected individuals. Thromb Haemost 2010;104(6):1228-1234. [http://dx.doi.org/10.1160%2FTH10-04-0233] [ Links ]
Accepted 26 January 2012.
Corresponding author: J Opie (jessica.opie@uct.ac.za)


{kind=link}
{kind=link}