










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSAMJ: South African Medical Journal
On-line version ISSN 2078-5135
Print version ISSN 0256-9574
SAMJ, S. Afr. med. j. vol.102 n.6 Pretoria Jun. 2012
FORUM
REVIEW
Approaches to target identification and validation for tuberculosis drug discovery: A University of Cape Town perspective
Digby F WarnerI; Valerie MizrahiII
ISenior research officer in the MRC/NHLS/UCTMolecular Mycobacteriology Research Unit, DST/NRF Centre of Excellence for Biomedical TB Research, Institute of Infectious Disease and Molecular Medicine, University of Cape Town (UCT)
IIDirector of the Institute of Infectious Disease and Molecular Medicine, UCT
ABSTRACT
Tuberculosis (TB) disproportionately affects a few high-burden countries including South Africa. In these regions, basic TB research is rare, endemic countries being valued primarily as sites for drug trials and clinical studies. Our basic mycobacterial research focuses on current approaches to drug target identification and validation within the context of international trends in TB drug discovery. Increased funding for TB drug development globally prompted a significant shift in the composition of drug discovery consortia, with academic laboratories assuming a major role in collaboration with industrial partners. This hybrid model holds promise for the expansion of local programmes, especially where actively supported by government. However, the application of industry-standard business practices to research projects involving biology and chemistry expertise demands a greater appreciation of the differences between a chemically, versus biologically, validated drug target, and of the factors informing these differences.
The strategic decision to transfer the Molecular Mycobacteriology Research Unit (MMRU) to the Faculty of Health Sciences (FHS) of the University of Cape Town (UCT) was made in January 2011. Established in 2000, the MMRU was originally a joint unit of the Medical Research Council (MRC), the South African Institute of Medical Research (SAIMR) and the University of the Witwatersrand (Wits), becoming a joint venture of the National Health Laboratory Service (NHLS), the MRC and Wits when the SAIMR was absorbed into the NHLS. The MMRU laboratory research of the physiology and metabolism of Mycobacterium tuberculosis (MTB) relevant to tuberculosis (TB) drug discovery is of particular significance for one of us (VM) - a former graduate of the Chemistry Department of UCT and currently Director of the Institute of Infectious Disease and Molecular Medicine (IIDMM). The IIDMM is a trans-faculty institute based physically and administratively in the FHS with staff from both the FHS and the Faculty of Science. The IIDMM provides an ideal framework for a research programme focused on TB drug discovery, with integrated chemistry and biology. It offers an opportunity to train a new generation of postgraduate students who can work at the interface of chemistry and medical microbiology which, historically, have been separated along faculty lines. A chemistry student whose research explores the structure-activity relationships within a compound series with demonstrated anti-mycobacterial activity would benefit from a deeper understanding of the tubercle bacillus, and the challenges of working with this extremely slow-growing pathogen under biosafety level 3 containment. Conversely, by engaging actively with medicinal chemists, the mycobacteriology student learns to exploit the power of using small-molecule inhibitors to probe the biology of MTB, and to identify and validate drug targets. These skills will be critical to overcoming the significant barriers to TB drug discovery.
With its exceptional strength in epidemiological and clinical TB research, South Africa (SA) has become the leading site in the world for evaluating new TB drug candidates developed elsewhere.1 Our long-term vision, which aligns with the Ten Year Innovation Plan of the Department of Science and Technology,2 is for SA to become a significant contributor along the entire TB drug-discovery and development pipeline. We look forward to developing a research programme at UCT that contributes meaningfully towards realising this vision.
TB in Africa
TB is a global problem. One-third of the world's population is infected, with recent reports estimating around 9 million new TB cases and 1.8 million deaths per annum.3 These numbers disguise a massive disparity in disease burden: 80% of all cases occurred in 22 countries, 9 of which are African. Africa accounted for 2.8 million cases and 430 000 deaths. SA ranks third in TB burden, behind India and China, and contributes 20% of all human immunodeficiency virus (HIV)-positive incident cases.4
The widely administered bacillus Calmette-Guerin (BCG) vaccine offers no protection against adult TB. Control efforts focus on treatment of active disease but are undermined by factors including HIV, extended duration of combination therapies requiring a minimum 6-month treatment period, increasing emergence of multidrug-resistant (MDR) and extensively drug-resistant (XDR) strains that require longer and more specialised treatment regimens, prevalence of other chronic diseases, diagnostic delays, and numerous social and economic confounders. New drugs which are safer, shorter-acting, and can be used in the context of other chronic illnesses, are crucially needed.5
New drug discovery for TB
Most frontline drugs were developed 50 years ago and were either not designed for the purpose or were supported by limited clinical trial data upon introduction. Regimens evolved empirically and remain non-optimal. The effective restriction of the TB burden to developing economies, and the misplaced confidence in the power of antibiotics to eliminate infectious disease, led to neglect in new drug development. However, the emergence of HIV and MTB drug resistance have fuelled the resurgence of TB as a global threat demanding renewed commitment to the discovery and development of new TB drugs.5
Major philanthropic partnerships and initiatives are now devoted to fighting the disease. Investment in TB research increased to more than 600 million US dollars by 2009 and enabled the formation of large, multidisciplinary research consortia, which has had a significant impact on the development, conduct and evaluation of TB drug discovery. Projects are ambitious and require the participation of multiple collaborative partners spanning academia, small biotechnology companies, and industry. In turn, this necessitates a more dispassionate assessment of project viability. The goal is inhibition of a validated target with new chemical entities that fulfil stringent pharmacological endpoints; there is no longer room for an academic researcher's sentimental attachment to an interesting intellectual problem which fails to satisfy key requirements for progression through the drug-development pipeline.
What should a new TB drug offer?
New-generation TB drugs must satisfy onerous criteria that should:5 shorten the duration of chemotherapy, either as a component of a new combination, or as an adjunct to existing drugs; be associated with minimal side-effects as part of a regimen that requires fewer tablets and less frequent dosing; possess activity against MDR and XDR strains; be compatible with other drugs for chronic conditions, especially HIV and diabetes, which can significantly impact on disease outcomes; and be active against the organism in both intra-and extracellular host environments. With the possible exception of drug-drug interactions, the difficulties associated with tackling TB chemotherapeutically are a direct consequence of the inherent characteristics of MTB and TB disease pathology. Therefore, efforts to improve therapeutic efficacy depend critically on an improved understanding of the physiology of the infecting organism.6
Mycobacterial physiology and drug discovery
The persistence of human MTB depends on its ability to cycle through repetitive phases of infection, disease, transmission, and clinical latency. Research into the fundamental physiology of MTB promises to reveal critical vulnerabilities in each phase that might be exploited for new drug discovery. However, fundamental gaps in our knowledge of basic biology and of the complexities of the host-pathogen interaction ensure that the translation of experimental observations into practical interventions remains challenging.7
MTB, an obligate pathogen well equipped to survive variable and hostile environments, is a formidable adversary. It has an expanded metabolic repertoire and a complex, impermeable cell wall that ensures resistance to host immune effectors and many antibiotic agents, while an array of transcriptional regulators enables a rapid response to environmental cues. The bacillus is also armed with multiple defence and detoxification pathways and DNA repair and maintenance functions8 and has adapted exquisitely to host colonisation.9 This is crucial to understanding the difficulties associated with new drug development for TB, and underlies the disconnection between our knowledge of mycobacterial physiology and observed pathogenesis. It remains difficult to link clinical phenotypes to an underlying bacterial or physiological state, and even more difficult to model these. Compounding this is limited understanding of bacterial metabolism during host infection10 and the fact that the functions of approximately 50% of the genes in its genome remain unknown11 (more than 10 years after the first MTB strain was sequenced).
Properties of a good drug target
Amalgamating expertise in chemistry and biology in drug discovery and development has resulted in redefined criteria for target selection to consider biological and pharmacological properties. The target should be essential for growth or survival of the organism under a chosen condition - so-called essentiality - and should be amenable to chemical inhibition - a property commonly referred to as druggability - and should be accessible to the inhibiting agent. Recognising that the attribute essentiality does not guarantee the attractiveness of a target is a reflection of the increasing sophistication of TB drug-discovery efforts. In turn, it calls for a re-evaluation of the role of the biology and chemistry components of drug discovery in target identification and validation.
Traditionally, target identification and validation has included activities designed to identify reactions or pathways that are critical for aspects of pathogenesis (primarily bacterial viability and/ or virulence), and the subsequent demonstration, genetically or chemically, that disruption of the reaction or pathway significantly impairs the identified property under disease-relevant conditions. This approach fails to separate the attributes of a validated target, as assessed pharmacologically/chemically, from the biological properties of a vulnerable protein/pathway inferred from molecular genetics (Fig. 1). From a practical perspective, this separation is critical since it informs the distinct - but overlapping - functions of the chemistry and biology teams in any TB drug-discovery chain.
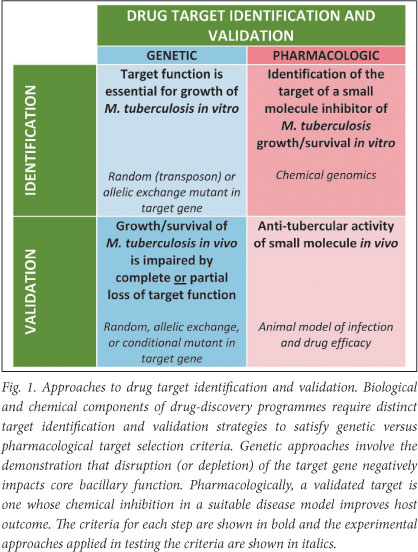
Target identification
In the early 2000s, Sassetti and colleagues12 developed a random mutagenesis approach that has been successfully applied to identify genes whose disruption severely attenuates the proliferative ability or virulence of MTB under specific conditions.13 These essential gene lists have been instrumental in elucidating multiple potential drug targets.
Because essentiality dominates target identification algorithms, many studies have yielded useful target identification (and validation) data in the absence of a drug-targeting motive, e.g. by demonstrating essential gene function in a specific disease model. However, perhaps less obvious is the critical contribution of fundamental research in mycobacterial physiology to eliminate an otherwise attractive target, owing to the unexpected identification of complementary or alternative pathway function. This applies in particular to central metabolism,10 where incomplete knowledge of the metabolic states adopted by MTB during host infection plagues predictions of essential pathways. Even where a specific metabolite (or cofactor) is known to be crucial to the infection cycle, key questions remain: How many pathways are available to the organism to synthesise the particular metabolite? Can the organism scavenge the metabolite (or precursors) from the host? Is the metabolite required throughout the course of infection, or only in certain disease states? Future efforts will demand the use of additional techniques such as in vitro and in vivo imaging,7 bacterial cell content (metabolite) profiling,14 systems biology,15 and metabolic modelling,16 designed to gain insight into the essentiality of a specific target that genetic analyses alone cannot provide.
Target identification increasingly involves the elucidation of the target(s) of compounds with demonstrated antibacterial activity, but whose mode of action is unknown. This is a critical component of modern drug-discovery programmes which are designed to identify lead molecules/compound classes with whole-cell activity from screens of compound and natural product libraries. Here, the goal of target identification and validation is effectively one of reverse engineering: the target of a compound with demonstrated inhibitory capacity is identified by means of chemical genomics - a combination of techniques that includes the isolation and sequencing of resistant mutants, whole genome transcriptional profiling, and assays of macromolecular synthesis (RNA, protein, DNA, peptidoglycan, and fatty acids) - and then validated through targeted gene modification or chemical genetic approaches. While the failure to identify the target does not necessarily disqualify a compound as a candidate chemical scaffold, it complicates subsequent efforts toward enhancement of the pharmacological properties of the active compound.
When is a target validated?
Target validation requires the demonstration in vivo that abrogation of function of a specific gene product results in a loss of bacterial viability (or phenotypic alteration) that is sufficiently dramatic to suggest that gene product as an attractive target for antimicrobial compounds (Fig. 1). Genetic approaches that rely on introducing mutations in genes in the MTB genome, in either a targeted or random fashion, are limited as abrogation of function of the mutated gene is permanent. Although this might not always impact on an observed phenotype, there are instances - especially where in vivo models are used - in which a greater level of control is required to identify the disease stage-specific contribution of a gene product to bacterial pathogenesis. The development of systems for the conditional regulation of gene expression in MTB under in vitro and in vivo conditions offers an elegant solution to this problem, provides unprecedented insights into otherwise intractable metabolic pathways, and enables the simultaneous assessment of multiple conditional mutants in a single experiment17 - a modification that holds promise for the development of high-throughput in vivo screens.
By definition, conditional expression systems depend on the ability to titrate the expression of the target gene in response to a particular stimulus. Therefore, it can be expected that for an essential gene (or antibiotic target), a critical threshold must exist below which a particular phenotype (growth retardation, loss of cell-wall integrity, etc.) will be manifest. Moreover, it is likely that this threshold differs for different targets. In turn, this suggests the potential application of regulated expression systems to the question of target vulnerability, which can be defined as the degree of inhibition of the target's function, such as enzymatic activity, that is required to impact cellular function detrimentally.18 The possibility of prioritising according to vulnerability could, therefore, ensure the allocation of maximum resources to the most tractable targets. A recent system for regulated protein degradation18 provided convincing evidence that the level of inhibition required to impact cellular function is target-specific, reinforcing the idea that target vulnerability might offer a more sophisticated measure in the validation process.
What about the future? TB drug discovery in an endemic country
The 12-year period since the inception of the MMRU spans the most dramatic era in the modern history of TB drug discovery and development. From an early focus on fundamental mycobacterial metabolism, the unit's research agenda shifted to the study of metabolic processes within a drug-discovery framework. The central motivation for our research remains to understand (and counteract) the disease caused by MTB, for which it is necessary to describe key pathways defining mycobacterial physiology and metabolism.8 Until recently, our projects were not undertaken with target identification and validation as primary outcomes, and instead aimed to establish the capacity of the organism to perform a specific metabolic function, and then to elucidate the contributing genetic components. From a discovery perspective, the mutant strains of MTB that were generated during the course of our studies offered the possibility of being applied as biological probes in specific disease models. More recently, the increased impetus to identify targets, and specific inhibitors of those targets, suggests a similar possibility; screens are likely to yield pools of compounds that fail to progress through the development pipeline but which might fulfil a similar function as chemical probes.19
We will continue to exploit molecular techniques that offer the best prospect of satisfying our dual mandate: to inform TB drug discovery, and to reveal metabolic functions that might contribute to pathogenesis. By integrating this approach with an established medicinal chemistry platform at UCT, we aim to expedite this process, and ensure that a South African laboratory contributes to this long-neglected problem.
Acknowledgements
Research in the MMRU is supported by the Department of Science and Technology, the National Research Foundation, the South African Medical Research Council, the National Health Laboratory Service and the University of Cape Town.
References
1. Diacon AH, Pym A, Grobusch M, et al The diarylquinoline TMC207 for multidrug-resistant tuberculosis. N Engl J Med 2009;360(23):2397-2405. [http://dx.doi.org/10.1056/NEJMoa0808427] [ Links ]
2. http://www.dst.gov.za/publications-policies/strategies-reports/The%20Ten-Year%20Plan%20for%20Science%20and%20Technology.pdf (accessed February 2012). [ Links ]
3. Dye C. Global epidemiology of tuberculosis. Lancet 2006;367(9514):938-940. [http://dx.doi.org/10.1016/S0140-6736(06)68384-0] [ Links ]
4. World Health Organization (WHO). Global Tuberculosis Control - Surveillance, Planning, Financing Geneva: WHO, 2010. [ Links ]
5. Koul A, Arnoult E, Lounis N, Guillemont J, Andries K. The challenge of new drug discovery for tuberculosis. Nature 2011;469(7331):483-490. [http://dx.doi.org/10.1038/nature09657] [ Links ]
6. Barry CE 3rd, Boshoff HI, Dartois V, et al. The spectrum of latent tuberculosis: rethinking the biology and intervention strategies. Nat Rev Microbiol 2009;7(12):845-855. [http://dx.doi.org/10.1038/nrmicro2236] [ Links ]
7. Russell DG, Barry CE 3rd, Flynn JL. Tuberculosis: what we don't know can, and does, hurt us. Science 2010;328(5980):852-826. [http://dx.doi.org/10.1126/science.1184784] [ Links ]
8. Warner DF, Mizrahi V Tuberculosis chemotherapy: the influence of bacillary stress and damage response pathways on drug efficacy. Clin Microbiol Rev 2006;19(3):558-570. [http://dx.doi.org/10.1128/CMR.00060-05]
9. Djelouadji Z, Raoult D, Drancourt M. Palaeogenomics of Mycobacterium tuberculosis: epidemic bursts with a degrading genome. Lancet Infect Dis 2011;11(8):641-650. [http://dx.doi.org/10.1016/S1473-3099(11)70093-7] [ Links ]
10. Rhee KY, Carvalho LP, Bryk R, et al. Central carbon metabolism in Mycobacterium tuberculosis: an unexpected frontier. Trends Microbiol 2011;19(7):307-314. [http://dx.doi.org/10.1016/j.tim.2011.03.008] [ Links ]
11. Lew JM, Kapopoulou A, Jones LM, Cole ST. TubercuList - 10 years after. Tuberculosis (Edinb) 2011;91(1):1-7. [http://dx.doi.org/10.1016/j.tube.2010.09.008] [ Links ]
12. Sassetti CM, Boyd DH, Rubin EJ. Comprehensive identification of conditionally essential genes in mycobacteria. Proc Natl Acad Sci USA 2011;98(22):12712-12717. [http://dx.doi.org/10.1073/pnas.231275498] [ Links ]
13. Griffin JE, Gawronski JD, Dejesus MA, Ioerger TR, Akerley BJ, Sassetti CM. High-resolution phenotypic profiling defines genes essential for mycobacterial growth and cholesterol catabolism. PLoS Pathog 2011;7(9):e1002251. [http://dx.doi.org/10.1371/journal.ppat.1002251] [ Links ]
14. De Carvalho LP, Fischer SM, Marrero J, Nathan C, Ehrt S, Rhee KY. Metabolomics of Mycobacterium tuberculosis reveals compartmentalized co-catabolism of carbon substrates. Chem Biol 2010;17(10):1122-1131. [http://dx.doi.org/10.1016/j.chembiol.2010.08.009] [ Links ]
15. Stumpf MP, Robertson BD, Duncan K, Young DB. Systems biology and its impact on anti-infective drug development. Prog Drug Res 2007;64(1):3-20. [http://dx.doi.org/10.1007/978-3-7643-7567-6_1] [ Links ]
16. Beste DJ, McFadden J. System-level strategies for studying the metabolism of Mycobacterium tuberculosis. Mol Biosyst 2010;6(12):2363-2372. [http://dx.doi.org/10.1039/c003757p] [ Links ]
17. Blumenthal A, Trujillo C, Ehrt S, Schnappinger D. Simultaneous analysis of multiple Mycobacterium tuberculosis knockdown mutants in vitro and in vivo. PLoS One 2010;5(12):e15667. [http://dx.doi.org/10.1371/journal.pone.0015667] [ Links ]
18. Wei JR, Krishnamoorthy V, Murphy K, et al. Depletion of antibiotic targets has widely varying effects on growth. Proc Natl Acad Sci USA 2011;108(10):4176-4181. [http://dx.doi.org/10.1073/pnas.1018301108] [ Links ]
19. Falconer SB, Czarny TL, Brown ED. Antibiotics as probes of biological complexity. Nat Chem Biol 2011;17:415-423. [http://dx.doi.org/10.1038/nchembio.590] [ Links ]
Accepted 31 January 2012.
Corresponding author: V Mizrahi (valerie.mizrahi@uct.ac.za)

