










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSAMJ: South African Medical Journal
On-line version ISSN 2078-5135
Print version ISSN 0256-9574
SAMJ, S. Afr. med. j. vol.101 n.12 Pretoria Dec. 2011
GUIDELINE
Guideline for the treatment of myelodysplastic syndromes (MDS) in South Africa
V J Louw; F Bassa; S W Chan; L Dreosti; M du Toit; M Ferreira; K Gartrell; K Gunther; V Jogessar; N Littleton; J Mahlangu; A McDonald; M Patel; R Pool; P Ruff; A Schmidt; G Sissolak; A Swart; E Verburgh; M J Webb
ABSTRACT
INTRODUCTION: Myelodysplastic syndromes (MDS) encompass a heterogeneous group of clonal haematopoietic disorders characterised by chronic and progressive cytopenias resulting from ineffective haematopoiesis. Treatment is complicated by differences in disease mechanisms in different subgroups, variable clinical phenotypes and risk of progression to acute myeloid leukaemia.
RATIONALE: Changes in disease classification, prognostic scoring systems, the availability of novel treatment options and the absence of South African guidelines for the diagnosis and management of these complex disorders underpinned the need for the development of these recommendations.
METHODS: These recommendations are based on the opinion of a number of experts in the field from the laboratory as well as clinical settings and came from both the private and institutional academic environments. The most recent literature as well as available guidelines from other countries were discussed and debated at a number of different meetings held over a 2-year period.
RESULTS: A comprehensive set of recommendations was developed focusing on risk stratification, supportive management and specific treatment. Novel agents and their indications are discussed and recommendations are made based on best available evidence and taking into account the availability of treatments in South Africa.
CONCLUSION: Correct diagnosis, risk stratification and appropriate therapeutic choices are the cornerstones of success in the management of patients with MDS.
1. Introduction
Myelodysplastic syndromes (MDS) encompass a heterogeneous group of clonal haematopoietic disorders characterised by chronic and progressive cytopenias resulting from ineffective haematopoiesis. Clinically, patients present with symptomatic anaemia, recurrent infections and bleeding. The morphological hallmarks of MDS are progressive dysplastic features of haematopoietic cells at all stages of development in blood and bone marrow. In many patients, depending on a number of prognostic factors, MDS transforms over time to acute myeloid leukaemia (AML). The latter is usually highly resistant to therapy and responses, if they occur, are usually short-lived. Treatment of symptomatic anaemia with frequent blood transfusions over protracted periods of time puts patients at risk of the effects of iron overload.
In the past decade, our understanding of the pathogenesis of MDS has improved greatly. Furthermore, the classification of MDS into different clinicopathological sub-groups is undergoing constant change and novel treatments are increasing our therapeutic options. In addition to the use of allogeneic stem cell transplantation, high-dose chemotherapy and growth factor support, a number of new drugs were recently approved by the US Food and Drug Administration (FDA) and the European Medicine Authority (EMEA) in Europe for patients with MDS. These include lenalidomide (currently awaiting Medicines Control Council (MCC) approval in South Africa and available only through named-patient section 21 MCC application) and two inhibitors of DNA methyltransferase (hypomethylating agents), azacitidine and decitabine. Decitabine has not been registered in South Africa yet, but is also available on a named-patient section 21 application basis.
Although these products were originally approved on the basis of response rates, transfusion requirements and improvements in quality of life, survival data were lacking. This changed with evidence of improved overall survival and an increase in the time to progression to AML with the demethylating agent azacitidine.1,2 These benefits were apparent even in subgroups with unfavourable karyotypes. The data were recently confirmed in the large, prospective, international, multicentre, open-label, randomised phase III AZA 001 trial,3 and have led to changes in international guidelines, such as the US-based National Comprehensive Cancer Network (NCCN), which now include azacitidine in their therapeutic algorithms.
2. Limitations of the guidelines
The recommendations represent a consensus view on reasonable methods of management applicable to most patients, but do not exclude other reasonable management options, and success cannot be guaranteed in every situation. The unique circumstances of each patient should be taken into account by the responsible specialist regarding decisions on any specific therapy.
3. Objective
Our objective was to review the National Comprehensive Cancer Network (NCCN) 2009, 2010 and 2011 practice guidelines for the different subsets of MDS and adapt or modify their application to the South African clinical setting in order to provide recommendations for MDS management for South Africa.
4. Methods
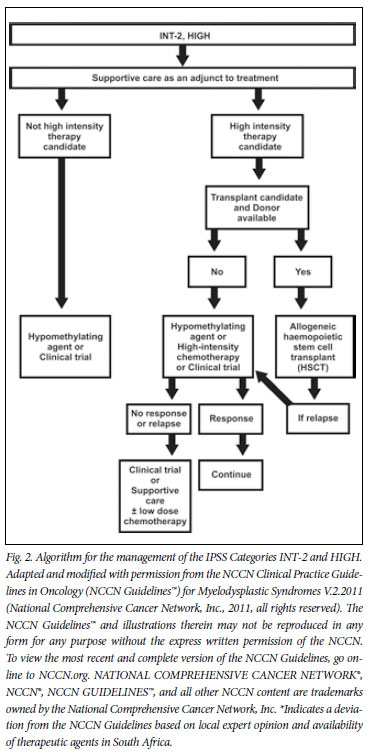
A panel of South African experts from the tertiary academic and private sectors met in Franschhoek, W Cape, in April 2009 to discuss contextualisation of the NCCN MDS clinical practice guidelines in the South African setting. After an introductory discussion by author VJL, the panel specifically reviewed the treatment options for patients in the lower-, intermediate- and higher-risk groups, according to the International Prognostic Scoring System (IPSS) categories LOW, intermediate-1 (INT-1) and HIGH (IPSS category HIGH, INT-2).4 The discussion focused, inter alia, on patients needing intensive therapy (including allogeneic stem cell transplantation), those needing less or lower-intensity therapy (defined as the use of low-intensity chemotherapy or biological response modifiers), supportive care, and the evaluation and treatment of disease-related symptomatic anaemia.
All discussions were recorded for later transcription and collated afterwards. A draft set of recommendations was presented by VJL at a follow-up meeting of an expanded panel consisting of clinical haematologists and oncologists at Kapama River Lodge in Limpopo, in July 2010. These draft recommendations took cognisance of literature published since the meeting in 2009, including the updated NCCN 2010 MDS Clinical Practice Guidelines, as well as data presented at the annual meetings of the American Society of Hematology (ASH) in December 2009 and the European Haematology Association in June 2010. Decisions were made on a consensus basis after active debate of the issues at hand. Subsequent changes based on the NCCN 2011 guidelines were included and discussed on an ad hoc basis telephonically and by e-mail among the authors.
The panel concluded that in each prognostic grouping, treatment options would be provided, which would allow tailoring of treatment in both the state and private sectors depending on the availability of treatments.
5. Results
5.1 Risk stratification
The IPSS categories were used in planning therapeutic options because they provide risk-based patient evaluation.4 The panel takes cognisance of the fact that the IPSS and the World Health Organization (WHO) Prognostic Scoring System (WPSS) are being revised, but at this stage the IPSS is still the most extensively used system in South Africa. It should also be noted that novel prognostic scoring systems based on cytogenetics, co-morbidities and flow cytometry have been developed, but are not widely used in the South African setting yet.5
Patients with clinically significant cytopenias are usually stratified into two major risk groups, namely:
• lower-risk patients - IPSS LOW/INT-1 categories; and
• higher-risk patients - IPSS INT-2/HIGH categories.
Table I summarises the prognostic variables and risk groups with regard to scoring and median survival in years, taking different age groups (<60 years, >60 years) into account.
5.2 Aim of treatment
In lower-risk patients, therapy is aimed at haematological improvement, whereas for patients with higher-risk disease, limiting disease progression and improving survival are considered most important. Therapeutic options for consideration include supportive care, lower-intensity therapy, high-intensity therapy, biological response modifiers, immunosuppressive therapies (ISTs), stem cell transplantation and/or a clinical trial.
6. Lower-risk patients
6.1 Supportive care
All patients should receive supportive care as an adjunct to treatment, which includes observation, monitoring, transfusion of blood and blood products, psychosocial support and attending to quality of life issues. Red blood cell (RBC) transfusions (for symptomatic anaemia), platelet transfusions (for severe thrombocytopenia or thrombocytopenic bleeding), antibiotics and antifibrinolytic agents are all frequently used as part of supportive care. In addition, iron chelation (to manage iron overload) and recombinant human erythropoietin (EPO) with or without human granulocyte colonystimulating factor (G-CSF) may be useful in selected patients.
6.2 RBC and platelet transfusions
Decision-making on transfusion need is similar to other indications, but as patients are often older with co-morbid conditions, the threshold for RBC transfusion may be higher (i.e. higher haemoglobin levels) and needs to be individualised to patients' symptoms and tolerance of chronic anaemia. As patients require recurrent transfusions, the use of pre-storage leukocyte-depleted blood is recommended to decrease the risks of platelet isosensitisation, viral infections, febrile transfusion reactions and immunosuppression. Platelets are generally only given to patients who are actively bleeding, except for those receiving active therapy (for example, allogeneic haematopoietic stem cell transplantation (HSCT), intensive chemotherapy, hypomethylating agents, IST), where prophylactic platelet transfusion may be appropriate. The use of universal irradiation of blood products is controversial, with the exception of patients considered for allogeneic HSCT.
6.3 Transfusion dependence and iron chelation
Iron overload is a frequent problem in chronically transfused patients. It has been shown that both transfusion dependency and elevated serum ferritin levels are associated with a decreased overall survival and possibly an increased risk for transformation to AML independently of cytogenetic risk groups.6-11 The effect on survival was more apparent in the lower-risk groups,10 in patients who were actively monitored for iron overload, and in patients undergoing HSCT.12,13 Iron chelation has been shown to significantly improve survival in heavily transfused patients in retrospective studies.14-17 In the prospective EPIC trial, deferasirox has been shown to be safe and effective in decreasing serum ferritin, alanine transaminase (ALT) and labile plasma iron (LPI) in 341 MDS patients with serum ferritin values >2 500 ng/ml.18,19 A recent consensus statement was published regarding the criteria for iron chelation in patients with transfusional iron overload in MDS (see Table II).20 The panel felt that these consensus guidelines can be applied in the South African setting. In patients whose haemoglobin normalises in response to treatment of their MDS, venesection is a reasonable alternative to consider for the management of iron overload. In most patients this will not be possible, and iron chelation with the oral iron chelator deferasirox, or continuous subcutaneous or intravenous infusions of deferrioxamine, should be considered. The ease of use of deferasirox makes this drug the iron chelator of choice in the setting of MDS. The aim should be to decrease serum ferritin levels to a target range of between 500 and 1 000 µg/l. Once the target range is achieved, the dosage of the iron chelator may be adjusted to maintain the patient within this range. A decrease in dose or interruption of the iron chelator is usually required once the serum ferritin level decreases below 500 µg/l.

It should be noted that, according to South African recommendations, deferasirox is contra-indicated in patients with high-risk MDS, in patients with a creatinine clearance <60 ml/min, and in patients with other haematological and non-haematological malignancies who are not expected to benefit from chelation therapy owing to rapid disease progression and limited life expectancy. Cases of acute renal failure, hepatic failure and fatal gastro-intestinal haemorrhage have been reported, with the latter occurring especially in elderly patients with advanced haematological malignancies and/ or low platelet counts. Most of the patients who developed these problems had co-morbid diseases that put them at risk for these complications. Table II summarises the criteria for iron chelation therapy in MDS, and which patients with MDS are most likely to benefit from iron chelation.
6.4 Management and prevention of infections
Infection, especially in neutropenic patients, is a major cause of death in patients with MDS and needs to be treated aggressively and appropriately. Broad-spectrum antibiotics are usually required for patients with neutropenic fever. The use of prophylactic antibiotics is less clear and they are not routinely recommended for all patients, but individual patients receiving treatments that put them at high risk for neutropenic fever may benefit from antibacterial and antifungal prophylaxis. This decision needs to be individualised taking into account the patient's clinical condition, intensity of treatment, neutropenic fever risk and co-morbid conditions. It should also be noted that iron overload has been associated with a higher risk of bacterial and fungal infections.21
7. Patients with symptomatic anaemia
Beyond the adjunctive measures of supportive care, patients are stratified according to whether they primarily present with symptomatic anaemia, thrombocytopenia or neutropenia. Symptomatic, transfusion-dependent anaemia is a common and important manifestation of MDS. It has been associated with reduced quality of life, iron overload and other complications of transfusions and the need for iron chelation therapy. Also, the cost of transfusion and iron chelation may be considerable.
7.1 Deletion 5q- ± other cytogenetic abnormalities
The NCCN guidelines recommend the use of lenalidomide in patients with del 5q. Lenalidomide has been shown to yield transfusion independence in 67% of lower-risk patients with del 5q and a median duration of response of more than 2 years.7 Cytogenetic responses were seen in 73% of patients (45% major and 28% minor), with grade 3 or 4 myelosuppression during the first 3 months as the most prominent adverse events.7,22,23 Although lenalidomide has been approved by the FDA, the EMEA has been concerned about a potential increased risk of progression to AML in patients with del 5q. Preliminary results from a number of prospective clinical trials seem to be reassuring.23-25
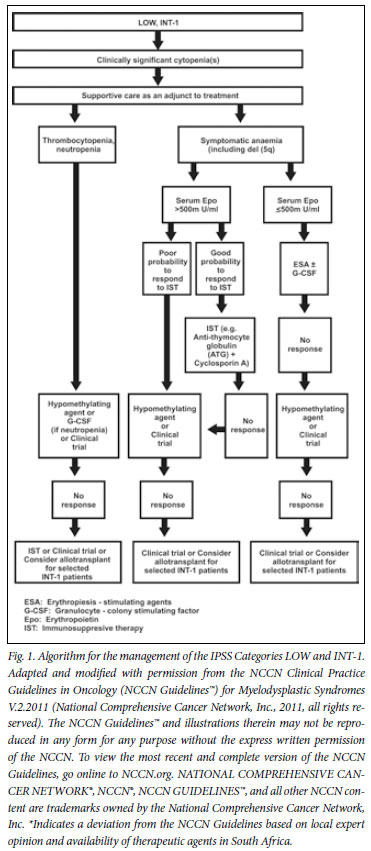
7.2 Erythropoiesis-stimulating agents ± G-CSF
A number of phase II clinical trials have shown response rates of 20 - 40% with single-agent erythropoiesis-stimulating agents (ESAs), with even higher response rates when treatment is instituted early.26 Combining ESAs with granulocyte-colony stimulating factor (G-CSF) may enhance response, owing to a synergistic effect.27-29 In a large cohort of 403 patients with MDS who received epoietin-alpha, epoietin-beta or darbepoietin with or without G-CSF, responses were seen in 62% of patients according to the International Working Group 200030 (IWG) criteria (40% major and 22% minor). Response duration (measured from onset rEPO) was 20 months (range 3 - 74 months).30 A meta-analysis of anaemic MDS patients confirmed the usefulness of epoietin-alpha and darbepoietin in the treatment of MDS.31
To manage these patients as cost-effectively as possible, the subsets of patients most likely to benefit need to be recognised.32 The strongest association with response to treatment has been seen in patients with EPO levels <500 mµ/ml and RBC transfusion needs.32 The serum EPO level cut-off point of 500 mU/ml (or U/l) was derived from two large phase II studies followed by validation in a prospective trial.29 The likelihood of response to EPO and G-CSF in a patient with a serum EPO level <500 mU/ml and a transfusion need of less than 2 units of RBCs per month was 74%. In contrast, patients with an EPO level >500 mU/ml and a transfusion need of >2 units per month only have a 7% chance of response.29 It is critical to identify these patients who are unlikely to respond, as they also have an increased possibility of disease progression, especially if other more appropriate therapies, such as allogeneic HSCT and azacitidine, are delayed.33
The effect of treatment on long-term outcomes has been favourable, but survival benefit was only seen in patients with a moderate pretreatment transfusion need of less than 2 units of RBCs per month.30,34,35 A recent study has further shown clear improvement in quality of life in responding patients that correlated with decreases in transfusion requirements.36 Most responses are seen within 12 weeks of treatment onset, with a median duration of response of about 2 years.30,34,37 The risk of progression to higher-risk MDS and AML does not seem to increase compared with patients treated with transfusion alone.30,34
The panel concluded that the stratification of low-risk patients into symptomatic anaemia, thrombocytopenia or neutropenia and according to serum EPO levels was valid, as was stratifying patients according to serum EPO levels. The use of ESAs in combination with G-CSF was supported in selected patients, taking into account the likelihood of response in an individual patient. In patients with a low likelihood of response, alternative therapies should be considered.
7.3 Failure to respond to ESA
In patients not responding to ESA and G-CSF, the available options in South Africa include hypomethylating agents, IST, a clinical trial or allogeneic HSCT in selected patients.
7.4 Anaemic patients with serum EPO level >500 mU/ml
These patients should be evaluated to determine whether they have a good probability of responding to IST, or whether they would be candidates for the use of a hypomethylating agent.
7.5 Immunosuppressive therapy
Patients most likely to respond to IST include IPSS LOW or INT-1 patients with one or more of the following features: <60 years old, HLA-DR15+, the presence of a paroxysmal nocturnal haemoglobinuria (PNH)-positive clone, or hypocellular bone marrow. Also, patients with a recent onset of RBC transfusion requirement, very few or no blasts and normal cytogenetics seem to respond better to IST.38-42 In these selected patients, antithymocyte globulin (ATG) in combination with cyclosporin should be considered, as responses can be expected in 30 - 40% of lower-risk patients resistant to EPOs. The panel concurred with the treatment pathway defined by the NCCN, i.e. that if patients are non-responders to IST, they would be considered for treatment with hypomethylating agents or a clinical trial.
Patients with a low probability of responding to IST should be considered for treatment with hypomethylating agents or a clinical trial. Once available, lenalidomide may become an appropriate option in selected patients. Patients not responding to any of these could be considered for allogeneic HSCT.
It is worth noting that very promising results have been seen in small studies with novel immunosuppressive regimens. In one study,43 alemtuzumab 10 mg/day was given for 10 days, with responses seen in 17 (77%) out of 22 patients with evaluable INT-1 disease, and in 4 (57%) out of 7 with evaluable INT-2 disease. Median time to response in this study was 3 months.43
7.6 Patients with predominant thrombocytopenia and/or neutropenia
In clinical practice, patients with true isolated neutropenia have shown very good responses to the addition of growth factors. As such, patients should be given a trial of growth factors or azacitidine, or be considered for a clinical trial. In the event of no or limited response, IST or allogeneic HSCT should be considered. The role and safety of romiplostim in MDS has not been fully delineated.
8. Higher-risk patients (IPSS INT-2/HIGH)
Therapeutic options for higher-risk patients include allogeneic HSCT, high-intensity therapy, low-dose chemotherapy (for example, low-dose cytarabine), hypomethylating agents, and an array of novel experimental agents or supportive care.
Patients in the INT-2 and HIGH risk categories according to the IPSS have a median survival of only 1.2 and 0.4 years, respectively. Time to AML progression is generally less than 1 year.4 Taking into account the very poor outcomes in this patient group, patients should be carefully evaluated and offered treatment that will positively change outcome and survival. Treatment for higher-risk patients is therefore dependent on whether they are felt to be candidates for intensive therapy, such as allogeneic HSCT or intensive chemotherapy.
Factors that influence this decision include patient age, performance status, absence of major co-morbid conditions, psychosocial status, and the availability of an HLA-matched donor. MDS is generally a disease of older patients who should be carefully evaluated for co-morbid disease that may influence their ability to tolerate intensive chemotherapy and/or HSCT. The HSCT co-morbidity index has been shown to be an accurate predictor of non-relapse mortality after HSCT.44-46 The panel agreed that the timing and selection of MDS patients for HSCT was critical. IPSS INT-2 and HIGH risk patients <60 years old should ideally proceed to allogeneic HSCT as early as possible. In LOW or INT-1 risk patients, HSCT is usually delayed until evidence of disease progression is observed. Non-myeloablative HSCT is usually preferred in order to reduce treatment-related mortality, except in younger patients, where full myeloablation is still often used.47 It has been suggested that potential transplant candidates may benefit from azacitidine pre-transplant during the period in which the donor search is ongoing, especially in patients with an unfavourable karyotype. Patients with unfavourable karyotypes are usually refractory to conventional chemotherapy. This concurs with the NCCN guidelines, that qualify the option of HSCT with the fact that azacitidine may be used as a bridge to transplant while awaiting improved patient status or donor availability. In patients with a more favourable karyotype, intensive chemotherapy may be useful to decrease blast percentages pre-transplant and thus limit post-transplant relapse rates.
8.1 High-intensity therapy
Intensive therapy is usually limited to patients less than 65 years of age with more favourable karyotypes and no allogeneic stem cell donor. In patients with unfavourable karyotypes, hypomethylating agents are preferred. In older patients, azacitidine is the treatment of choice,48 although careful attention to co-morbid disease is required. Anthracyclines and cytarabine combinations remain the preferred choice for intensive chemotherapy, and complete remission (CR) rates of between 40% and 60% are usually attained. Unfortunately, CR is not often sustained, with a median duration of CR of less than 12 months, with less than 10% of patients achieving a prolonged CR. Toxicity is often higher than that seen in patients treated for AML with similar regimens, and is often characterised by prolonged cytopenias due to bone marrow hypoplasia.49-51 Patients who generally benefit most from intensive chemotherapy are younger patients with favourable cytogenetics. Complete remission rates are low and CR duration is generally short in patients with an unfavourable karyotype.
In higher-risk patients, a clinical trial with an azacitidine-based combination may be considered before intensive treatment, especially in older patients. This is supported by data from the AZA 001 trial,3 a large, international, multicentre, open-label, randomised phase III trial. Findings demonstrated improved overall survival rates at 2 years (50.8% v. 26.2%) using azacitidine when compared with conventional care (best supportive care, intensive chemotherapy and low-dose cytarabine).3 This benefit in overall survival was independent of age, karyotype, the number of bone marrow blasts and the FAB or WHO type.3 Of note was the survival advantage seen in patients with poorrisk cytogenetics (-7/7q-) in all patient groups receiving azacitidine.48 It is important to emphasise that responses often occurred only after 4 - 6 cycles, with the median number of cycles being 9 and 14 given to non-responders and responders, respectively.3 In another analysis of the AZA 001 trial, azacitidine was shown to improve overall survival when compared with low-dose cytarabine, with fewer grade 3 - 4 cytopenias in the azacitidine group.48 A useful prognostic scoring system has been developed to predict responses to azacitidine.52
A second demethylating agent, decitabine, has not been found to confer a survival advantage in controlled trials, but this may at least in part have been due to the nature of the trials, in which therapy was limited to 4 cycles and a mixed population of MDS patients were treated.53,54 A more recent prospective, randomised phase III study55 showed that decitabine may lead to responses in older MDS patients, but without a significant improvement in overall and AMLfree survival. Transformation to AML was significantly decreased in this study. In this study, multivariate analysis showed that patients with a shorter duration of MDS had a worse outcome when given decitabine.55
Treatment options for patients who are not candidates for intensive therapy are similar to those in low-risk patients, being azacitidinebased regimens, supportive care, low-dose cytarabine or other experimental agents where available and appropriate. Older patients with poor-risk cytogenetics, who are not eligible for intensive chemotherapy, are usually offered an azacitidine-based therapy or best supportive care.
8.2 Non-intensive therapy
As a relatively low-cost intervention, low-dose cytarabine, given at a dose of 20 mg/m2/d for 14 -21 days out of every month, may yield partial and complete remission rates of 20% and 15%, respectively. These responses are generally limited to patients without poorrisk cytogenetics and are usually short-lived.48 Myelosuppression is very common, with grade 3 - 4 anaemia, neutropenia and thrombocytopenia seen in up to 77%, 89% and 96% of patients, respectively. Compared with azacitidine, response rates were fewer and shorter, overall survival lower and toxicity higher.48 Low-dose cytarabine could be considered as an adjunct to supportive care if azacitidine treatment has failed or is not available.
9. Experimental agents
It is generally felt that patients should be included in clinical trials as far as possible. Many new agents and drug combinations are being investigated. In patients with higher-risk MDS who are eligible for a transplant and have an available donor, allogeneic HSCT with or without a preceding hypomethylating agent is the treatment of choice. In all other patients, a hypomethylating agent would be the treatment of choice. In case of failure or loss of response after a hypomethylating agent, intensive chemotherapy or low-dose cytarabine are therapeutic options that may be considered. Supportive care should be offered to all patients while taking individual risk-benefit factors into account when making decisions on the use of more expensive treatment modalities, such as growth factors, EPOs and iron chelation.
10. Conclusion
The management of MDS is a complex process requiring expertise and skills in managing the complications of the disease as well as complications of treatment used. Continuous progress in the understanding of the molecular pathogenesis of MDS has led to the development of a number of therapeutic agents with acceptable side-effect profiles and improvements in survival and quality of life of MDS patients. The results from ongoing, prospective, randomised trials on novel agents and therapeutic combinations are eagerly awaited.
References
1. Silverman LR, Demakos EP, Peterson BL, et al. Randomized controlled trial of azacitidine in patients with the myelodysplastic syndrome: a study of the cancer and leukemia group B. J Clin Oncol 2002;20:2429-2440. [ Links ]
2. Kornblith AB, Herndon JE 2nd, Silverman LR, et al. Impact of azacytidine on the quality of life of patients with myelodysplastic syndrome treated in a randomized phase III trial: a Cancer and Leukemia Group B study. J Clin Oncol 2002;20:2441-2452. [ Links ]
3. Fenaux P, Mufti GJ, Hellstrom-Lindberg E, et al. Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: a randomised, open-label, phase III study. Lancet Oncol 2009;10:223-232. [ Links ]
4. Greenberg P, Cox C, LeBeau MM, et al. International scoring system for evaluating prognosis in myelodysplastic syndromes. Blood 1997;89:2079-2088. [ Links ]
5. Sperr WR, Wimazal F, Kundi M, et al. Comorbidity as prognostic variable in MDS: comparative evaluation of the HCT-CI and CCI in a core dataset of 419 patients of the Austrian MDS Study Group. Ann Oncol 2010;21:114-119. [ Links ]
6. Cazzola M, Malcovati L. Myelodysplastic syndromes - coping with ineffective hematopoiesis. N Engl J Med 2005;352:536-538. [ Links ]
7. List A, Kurtin S, Roe DJ, et al. Efficacy of lenalidomide in myelodysplastic syndromes. N Engl J Med 2005;352:549-557. [ Links ]
8. Malcovati L, Porta MG, Pascutto C, et al. Prognostic factors and life expectancy in myelodysplastic syndromes classified according to WHO criteria: a basis for clinical decision making. J Clin Oncol 2005;23:7594-7603. [ Links ]
9. Malcovati L, Della Porta MG, Cazzola M. Predicting survival and leukemic evolution in patients with myelodysplastic syndrome. Haematologica 2006;91:1588-1590. [ Links ]
10. Malcovati L. Impact of transfusion dependency and secondary iron overload on the survival of patients with myelodysplastic syndromes. Leuk Res 2007;31(Suppl 3):S2-6. [ Links ]
11. Sanz G, Nomdedeu B, Such E, et al. Independent impact of iron overload and transfusion dependency on survival and leukemic evolution in patients with myelodysplastic syndrome. ASH Annual Meeting Abstracts 2008;112:640. http://abstracts.hematologylibrary.org/cgi/content/abstract/112/11/640?maxtoshow=&hits=10&RESULTFORMAT=&fulltext=nomdedeu&searchid=1&FIRSTINDEX=0&volume=112&issue=11&resourcetype=HWCIT (accessed 13 September 2011). [ Links ]
12. Armand P, Kim HT, Cutler CS, et al. Prognostic impact of elevated pretransplantation serum ferritin in patients undergoing myeloablative stem cell transplantation. Blood 2007;109:4586-4588. [ Links ]
13. Ray LA, Wetzstein GA, Wang S-T, et al. Monitoring and treatment of transfusional iron overload: findings from an electronic medical records review study at the Moffitt Cancer Center and Research Institute. ASH Annual Meeting Abstracts 2010;116:1503. http://abstracts.hematologylibrary.org/cgi/content/abstract/116/21/1503?maxtoshow=&hits=10&RESULTFORMAT=&fulltext=wetzstein&searchid=1&FIRSTINDEX=0&volume=116&issue=21&resourcetype=HWCIT (accessed 13 September 2011). [ Links ]
14. Rose C, Brechignac S, Vassilief D, et al. Does iron chelation therapy improve survival in regularly transfused lower risk MDS patients? A multicenter study by the GFM (Groupe Francophone des Myelodysplasies). Leuk Res 2010;34:864-870. [ Links ]
15. Leitch HA. Improving clinical outcome in patients with myelodysplastic syndrome and iron overload using iron chelation therapy. Leuk Res 2007; 31(Suppl 3):S7-9. [ Links ]
16. Leitch H, Leger C, Goodman T, et al. Improved survival in patients with myelodysplastic syndrome receiving iron chelation therapy. Clinical Leukemia 2008;2:205-211. [ Links ]
17. Fox F, Kundgen A, Nachtkamp K, et al. Matched-pair analysis of 186 MDS patients receiving iron chelation therapy or transfusion therapy only. ASH Annual Meeting Abstracts 2009;114:1747. http://abstracts.hematologylibrary.org/cgi/content/abstract/114/22/1747?maxtoshow=&hits=10&RESULTFORMAT=&fulltext=kundgen&searchid=1&FIRSTINDEX=0&sortspec=relevance&resourcetype=HWCIT (accessed 13 September 2011). [ Links ]
18. Cappellini MD, Porter J, El-Beshlawy A, et al. Tailoring iron chelation by iron intake and serum ferritin: the prospective EPIC study of deferasirox in 1744 patients with transfusion-dependent anemias. Haematologica 2010;95:557-566. [ Links ]
19. Gattermann N, Finelli C, Porta DM, et al. Deferasirox in iron-overloaded patients with transfusiondependent myelodysplastic syndromes: Results from the large 1-year EPIC study. Leuk Res 2010;34(9):1143-1150. [ Links ]
20. Bennett JM. Consensus statement on iron overload in myelodysplastic syndromes. Am J Hematol 2008;83:858-861. [ Links ]
21. Mattiuzzi G, Amin HM, Kantarjian H, Garcia-Manero G, Cortes J. Baseline serum ferritin predicts rate of infection in patients with acute myelogenous leukemia and high-risk myelodysplastic syndrome. ASH Annual Meeting Abstracts 2009;114:1611. http://abstracts.hematologylibrary.org/cgi/content/abstract/114/22/1611?maxtoshow=&hits=10&RESULTFORMAT=1&author1=mattiuzzi&author2=amin&title=ferritin&andorexacttitle=and&andorexacttitleabs=and&andorexactfulltext=and&searchid=1&FIRSTINDEX=0&sortspec=relevance&fdate=1/1/2009&resourcetype=HWCIT (accessed 13 September 2011). [ Links ]
22. List A, Dewald G, Bennett J, et al. Lenalidomide in the myelodysplastic syndrome with chromosome 5q deletion. N Engl J Med 2006;355:1456-1465. [ Links ]
23. Fenaux P, Giagounidis A, Selleslag D, et al. RBC transfusion independence and safety profile of lenalidomide 5 or 10 mg in patients with LOW- or INT-1-risk MDS with del5q: results from a randomized phase III trial (MDS-004). ASH Annual Meeting Abstracts 2009;114:944. http://abstracts.hematologylibrary.org/cgi/content/abstract/114/22/944?maxtoshow=&hits=10&RESULTFORMAT=&fulltext=selleslag&searchid=1&FIRSTINDEX=0&sortspec=relevance&resourcetype=HWCIT (accessed 13 September 2011). [ Links ]
24. Germing U, Lauseker M, Hildebrandt B, et al. Survival, prognostic factors, and rates of leukemic transformation in a multicenter study of 303 untreated patients with MDS and del(5q). ASH Annual Meeting Abstracts 2009;114:945. http://abstracts.hematologylibrary.org/cgi/content/abstract/114/22/945?maxtoshow=&hits=10&RESULTFORMAT=&fulltext=lauseker&searchid=1&FIRSTINDEX=0&sortspec=relevance&resourcetype=HWCIT (accessed 13 September 2011). [ Links ]
25. Le Bras F, Sebert M, Kelaidi C, et al. Treatment of lower risk MDS with del 5q with lenalidomide (LEN): results of the french ATU program. ASH Annual Meeting Abstracts 2009;114:2764. http://abstracts.hematologylibrary.org/cgi/content/abstract/114/22/2764?maxtoshow=&hits=10&RESULTFORMAT=&fulltext=kelaidi&searchid=1&FIRSTINDEX=0&sortspec=relevance&resourcetype=HWCIT (accessed 13 September 2011). [ Links ]
26. Hellstrom-Lindberg E, Malcovati L. Supportive care and use of hematopoietic growth factors in myelodysplastic syndromes. Semin Hematol 2008;45:14-22. [ Links ]
27. Jadersten M, Montgomery SM, Dybedal I, Porwit-MacDonald A, Hellstrom-Lindberg E. Long-term outcome of treatment of anemia in MDS with erythropoietin and G-CSF. Blood 2005;106:803-811. [ Links ]
28. Balleari E, Rossi E, Clavio M, et al. Erythropoietin plus granulocyte colony-stimulating factor is better than erythropoietin alone to treat anemia in low-risk myelodysplastic syndromes: results from a randomized single-centre study. Ann Hematol 2006;85:174-180. [ Links ]
29. Hellstrom-Lindberg E, Gulbrandsen N, Lindberg G, et al. A validated decision model for treating the anaemia of myelodysplastic syndromes with erythropoietin + granulocyte colony-stimulating factor: significant effects on quality of life. Br J Haematol 2003;120:1037-1046. [ Links ]
30. Park S, Grabar S, Kelaidi C, et al. Predictive factors of response and survival in myelodysplastic syndrome treated with erythropoietin and G-CSF: the GFM experience. Blood 2008;111:574-582. [ Links ]
31. Moyo V, Lefebvre P, Duh MS, Yektashenas B, Mundle S. Erythropoiesis-stimulating agents in the treatment of anemia in myelodysplastic syndromes: a meta-analysis. Ann Hematol 2008;87:527-536. [ Links ]
32. Hellstrom-Lindberg E. Erythropoiesis-stimulating agents in myelodysplastic syndromes. Leuk Lymphoma 2010;51:1155-1156. [ Links ]
33. Malcovati L, Germing U, Kuendgen A, et al. Time-dependent prognostic scoring system for predicting survival and leukemic evolution in myelodysplastic syndromes. J Clin Oncol 2007;25:3503-3510. [ Links ]
34. Balleari E, Clavio M, Arboscello E, et al. Weekly standard doses of rh-EPO are highly effective for the treatment of anemic patients with low-intermediate 1 risk myelodysplastic syndromes. Leuk Res 2011;Jul 25. [Epub ahead of print] [ Links ].
35. Greenberg PL, Sun Z, Miller KB, et al. Treatment of myelodysplastic syndrome patients with erythropoietin with or without granulocyte colony-stimulating factor: results of a prospective randomized phase 3 trial by the Eastern Cooperative Oncology Group (E1996). Blood 2009;114:23932400. [ Links ]
36. Oliva EN, Nobile F, Alimena G, et al. Darbepoetin alfa for the treatment of anemia associated with myelodysplastic syndromes: efficacy and quality of life. Leuk Lymphoma 2010;51:1007-1014. [ Links ]
37. Jadersten M, Malcovati L, Dybedal I, et al. Erythropoietin and granulocyte-colony stimulating factor treatment associated with improved survival in myelodysplastic syndrome. J Clin Oncol 2008;26:36073613. [ Links ]
38. Lim ZY, Killick S, Germing U, et al. Low IPSS score and bone marrow hypocellularity in MDS patients predict hematological responses to antithymocyte globulin. Leukemia 2007;21:1436-1441. [ Links ]
39. Steensma DP, Dispenzieri A, Moore SB, Schroeder G, Tefferi A. Antithymocyte globulin has limited efficacy and substantial toxicity in unselected anemic patients with myelodysplastic syndrome. Blood 2003;101:2156-2158. [ Links ]
40. Saunthararajah Y, Nakamura R, Wesley R, Wang QJ, Barrett AJ. A simple method to predict response to immunosuppressive therapy in patients with myelodysplastic syndrome. Blood 2003;102:3025-3027. [ Links ]
41. Sloand EM, Wu CO, Greenberg P, Young N, Barrett J. Factors affecting response and survival in patients with myelodysplasia treated with immunosuppressive therapy. J Clin Oncol 2008;26:2505-2511. [ Links ]
42. Stadler M, Germing U, Kliche KO, et al. A prospective, randomised, phase II study of horse antithymocyte globulin vs rabbit antithymocyte globulin as immune-modulating therapy in patients with low-risk myelodysplastic syndromes. Leukemia 2004;18:460-465. [ Links ]
43. Sloand EM, Olnes MJ, Shenoy A, et al. Alemtuzumab treatment of intermediate-1 myelodysplasia patients is associated with sustained improvement in blood counts and cytogenetic remissions. J Clin Oncol 2010;28:5166-5173. [ Links ]
44. Giles FJ, Borthakur G, Ravandi F, et al. The haematopoietic cell transplantation comorbidity index score is predictive of early death and survival in patients over 60 years of age receiving induction therapy for acute myeloid leukaemia. Br J Haematol 2007;136:624-627. [ Links ]
45. Sorror ML, Sandmaier BM, Storer BE, et al. Comorbidity and disease status based risk stratification of outcomes among patients with acute myeloid leukemia or myelodysplasia receiving allogeneic hematopoietic cell transplantation. J Clin Oncol 2007;25:4246-4254. [ Links ]
46. Sorror ML, Maris MB, Storb R, et al. Hematopoietic cell transplantation (HCT)-specific comorbidity index: a new tool for risk assessment before allogeneic HCT. Blood 2005;106:2912-2919. [ Links ]
47. Martino R, Iacobelli S, Brand R, et al. Retrospective comparison of reduced-intensity conditioning and conventional high-dose conditioning for allogeneic hematopoietic stem cell transplantation using HLA-identical sibling donors in myelodysplastic syndromes. Blood 2006;108:836-846. [ Links ]
48. Fenaux P, Gattermann N, Seymour JF, et al. Prolonged survival with improved tolerability in higher-risk myelodysplastic syndromes: azacitidine compared with low dose ara-C. Br J Haematol 2010;149:244-249. [ Links ]
49. Gardin C, Turlure P, Fagot T, et al. Postremission treatment of elderly patients with acute myeloid leukemia in first complete remission after intensive induction chemotherapy: results of the multicenter randomized Acute Leukemia French Association (ALFA) 9803 trial. Blood 2007;109:5129-5135. [ Links ]
50. Beran M, Shen Y, Kantarjian H, et al. High-dose chemotherapy in high-risk myelodysplastic syndrome: covariate-adjusted comparison of five regimens. Cancer 2001;92:1999-2015. [ Links ]
51. Wattel E, De Botton S, Luc Lai J, et al. Long-term follow-up of de novo myelodysplastic syndromes treated with intensive chemotherapy: incidence of long-term survivors and outcome of partial responders. Br J Haematol 1997;98:983-991. [ Links ]
52. Itzykson R, Thepot S, Quesnel B, et al. Prognostic factors for response and overall survival in 282 patients with higher-risk myelodysplastic syndromes treated with azacitidine. Blood 2011;117:403-411. [ Links ]
53. Kantarjian H, Issa JP, Rosenfeld CS, et al. Decitabine improves patient outcomes in myelodysplastic syndromes: results of a phase III randomized study. Cancer 2006;106:1794-803. [ Links ]
54. WijerMans P, Suciu S, Baila L, et al. Low dose decitabine versus best supportive care in elderly patients with intermediate or high risk MDS not eligible for intensive chemotherapy: final results of the randomized phase III study (06011) of the EORTC Leukemia and German MDS Study Groups. ASH Annual Meeting Abstracts 2008;112:226. [ Links ]
55. Lubbert M, Suciu S, Baila L, et al. Low-dose decitabine versus best supportive care in elderly patients with intermediate- or high-risk myelodysplastic syndrome (MDS) ineligible for intensive chemotherapy: final results of the randomized phase III study of the European Organisation for Research and Treatment of Cancer Leukemia Group and the German MDS Study Group. J Clin Oncol 2011;29:1987-1996. [ Links ]
V J Louw, M J Webb, Clinical Haematology Unit, Department of Internal Medicine, University of the Free State, Bloemfontein;
F Bassa, G Sissolak, Division of Clinical Haematology, Department of Medicine, Tygerberg Hospital and Stellenbosch University, Tygerberg, W Cape;
S W Chan, P Ruff, Division of Medical Oncology, Faculty of Health Sciences, University of the Witwatersrand, Johannesburg;
L Dreosti, Department of Medical Oncology, University of Pretoria;
M du Toit, Cape Haematology Clinic and Bone Marrow Transplant Centre, Cape Town;
M Ferreira, Ampath Pathologists, Bloemfontein;
K Gartrell, Louis Leipoldt Medi-Clinic, Bellville, W Cape;
K Gunther, Mayo Centre/Life Flora Clinic, Johannesburg;
V Jogessar, Department of Haematology, University of KwaZulu-Natal and National Health Laboratory Service;
N Littleton, Department of Clinical Haematology, Provincial Hospital Port Elizabeth, E Cape;
J Mahlangu, Department of Molecular Medicine and Haematology, University of the Witwatersrand and National Health Laboratory Service, Johannesburg;
A McDonald, Division of Haematology, Groote Schuur Hospital and University of Cape Town;
M Patel, Clinical Haematology Unit, Department of Medicine, Chris Hani Baragwanath Hospital and Faculty of Health Sciences, University of the Witwatersrand;
R Pool, Department of Haematology, University of Pretoria;
A Schmidt, Oncology Department, Panorama Medical Centre, Panorama, W Cape;
A Swart, Department of Haematology and National Health Laboratory Service, University of Pretoria;
E Verburgh, Department of Haematology, St James's Hospital, Dublin, Ireland
Corresponding author: V Louw (louwvj.md@ufs.ac.za)


{kind=link}