










Servicios Personalizados
Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkSAMJ: South African Medical Journal
versión On-line ISSN 2078-5135
versión impresa ISSN 0256-9574
SAMJ, S. Afr. med. j. vol.101 no.10 Pretoria oct. 2011
ORIGINAL ARTICLES
A novel CYBB mutation with the first genetically confirmed case of chronic granulomatous disease in South Africa
R NaidooI; N JordaanVI; K W ChanVII; D M le RouxII; S PienaarIII; J NuttallIV; Y L LauVIII; B S EleyV
IMB ChB, FAAP. Paediatric Infectious Diseases Unit, Red Cross War Memorial Children's Hospital, and Department of Paediatrics and Child Health, University of Cape Town
IIMB ChB, FCP (Paed), MPH. Paediatric Infectious Diseases Unit, Red Cross War Memorial Children's Hospital, and Department of Paediatrics and Child Health, University of Cape Town
IIIMSc. Paediatric Infectious Diseases Unit, Red Cross War Memorial Children's Hospital, and Department of Paediatrics and Child Health, University of Cape Town
IVMB ChB, FCP (Paed). Paediatric Infectious Diseases Unit, Red Cross War Memorial Children's Hospital, and Department of Paediatrics and Child Health, University of Cape Town
VMB ChB, FCP (Paed). Paediatric Infectious Diseases Unit, Red Cross War Memorial Children's Hospital, and Department of Paediatrics and Child Health, University of Cape Town
VIMB ChB, MMed (Paed). Paediatrician in private practice, Cuyler Hospital, Uitenhage, Eastern Cape
VIIMPhil. Department of Paediatrics and Adolescent Medicine, University of Hong Kong, HKSAR, China
VIIIMD. Department of Paediatrics and Adolescent Medicine, University of Hong Kong, HKSAR, China
ABSTRACT
A case of a child with chronic granulomatous disease (CGD) presenting with recurrent mycobacterial infections and invasive Aspergillus fumigatus disease is described. Genetic analysis confirmed X-linked CGD with a novel mutation in exon 10 of the CYBB gene - the first South African report of genetically confirmed CGD.
Chronic granulomatous disease (CGD) is a rare primary immunodeficiency disease of phagocytic cells, resulting from impaired function of one of five essential subunits of the reduced nicotinamide adenine dinucleotide phosphate (NADPH) oxidase enzyme complex within neutrophils, eosinophils, monocytes and macrophages. This enzyme complex mediates intracellular killing of phagocytosed micro-organisms. The NADPH oxidase complex is composed of membrane-bound glycoproteins gp91phox and p22phox, and cytosolic subunits p47phox, p67phox, p40phox and rac. Although mutations in genes encoding any one of these protein subunits can result in the disease, 70% of cases are due to mutations in the CYBB gene causing gp91phox deficiency, which is responsible for X-linked CGD.1,2 Autosomal recessive forms of CGD due to mutations in genes encoding p47phox, p22phox, p67phox and p40phox have been described.2
Failure to mediate oxidative killing results in susceptibility to recurrent life-threatening bacterial and fungal infections and increased risk of mycobacterial infections.1 In the past, mortality from CGD was exceptionally high, but antibiotic and antifungal prophylaxis and therapy, and haematopoietic stem cell transplantation has improved outcomes significantly and reduced mortality to 5%.1 The worldwide prevalence of CGD is estimated to be 1 in 200 000 - 250 000 live births.1 Since 1983, 6 cases in South Africa have been reported in the literature, all diagnosed by immunological testing.3 We describe the first genetically confirmed case of CGD in this country.
Case report
In June 2010, a 5-year-old boy with a past history of recurrent tuberculosis (TB) presented to his paediatrician with left lower lobe consolidation, generalised lymphadenopathy and marked failure to thrive. Bacterial and mycobacterial cultures were negative and, in view of his past history, he was suspected of having drug-resistant TB. He had been well up until 15 months of age, when pulmonary TB was diagnosed, and he received 6 months of anti-TB medication. However, he presented a year later with abdominal TB. He responded to a second course of therapy, but at the age of 4 years presented with a discharging sinus over the right arm and right axillary scrofuloderma. Histological analysis of the skin biopsy confirmed the presence of intracellular acid-fast bacilli, although mycobacterial cultures were negative. He responded to a third course of anti-TB medication. There was no preceding history of severe acute bacterial infections.
Upon transfer to Red Cross War Memorial Children's Hospital, clinical evaluation revealed wasting, generalised lymphadenopathy, and left lower lobe consolidation on respiratory examination. There was tenderness over the thoracic spine but no gibbus formation. Although globally weak, no neurological abnormalities were present. Chest computed tomography (CT) showed opacification in the left lower lobe, multiple bilateral upper lobe parenchymal nodules, a contiguous left paravertebral density along thoracic vertebrae 7 - 11, erosion of ribs 7 and 8, and partial height loss of corresponding thoracic vertebral bodies. A lung biopsy showed granulomatous inflammation and septated hyphae with dichotomous branching resembling Aspergillus infection. Fungal cultures, however, were negative. He was initially commenced on amphotericin B but changed to intravenous voriconazole a week later.
The clinical course improved and he was converted to oral voriconazole after 3 weeks. However, a month into therapy, he developed collapse of thoracic vertebrae 6 -8 and required spinal cord decompression. Culture from the spinal mass confirmed Aspergillus fumigatus infection. The myelopathy improved gradually. However, 6 months later, mild bilateral lower limb hypertonia persisted. Follow-up chest CT showed calcification of the pulmonary nodules but persistence of the paravertebral mass and left lower lobe consolidation. The patient remains on long-term therapeutic oral voriconazole and cotrimoxazole prophylaxis.
Immunological work-up demonstrated evidence of reduced neutrophil killing capacity, i.e. a monocyte oxidative reaction of 14.26% (normal 70 - 100%) and neutrophil oxidative reaction of 17.44% (normal 95 - 100%), which suggested a diagnosis of CGD. This was confirmed on repeat neutrophil burst testing. Immunoglobulin concentrations, complement screen and lymphocyte subset analysis were relatively normal.
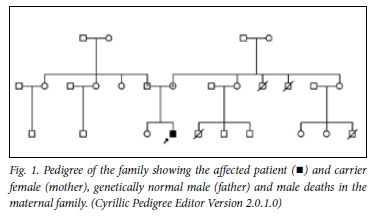
Although there was no reported family history of primary immunodeficiencies, 2 maternal uncles and 1 maternal male cousin died in infancy of unknown causes that suggested an X-linked inheritance pattern (Fig. 1). Sequencing of the CYBB gene on peripheral blood samples was completed by the University of Hong Kong;4 13 exon fragments were amplified using HotStarTaq Plus PCR system (Qiagen GmbH, Germany). Homology analysis of the sequenced data with CYBB genomic DNA was performed through the NCBI database (http://www.ncbi.nlm.nih.gov/BLAST) and the Ensembl SNPs database (http://www.ensembl.org). Analysis of the CYBB gene of the patient revealed a novel insertion-deletion mutation in exon 10, while his mother was found to be a heterozygous carrier of the mutation (Fig. 2).

Discussion
We describe a patient with CGD and a novel mutation in the CYBB gene. If the South African incidence is similar to international prevalence, 4 - 5 new cases of CGD should be identified in the country per year, which suggests that many children with CGD are not being diagnosed, most probably owing to failure to recognise the clinical manifestations.
Three mycobacterial infections and invasive aspergillosis in our patient is consistent with CGD. Acute bacterial infections with Staphylococcus aureus, Burkholderia cepacia, Serratia marcescens and Nocardia sp. predominate, while Aspergillus - the most frequently associated fungal infection - is the leading cause of morbidity and mortality.1,2 Several CGD patients with Bacille-Calmette-Guérin (BCG) disease and TB have been described in regions endemic for tuberculosis.4 Other CYBB mutations not related to CGD have also been associated with X-linked recessive mendelian susceptibility to mycobacteria.5 Other primary immunodeficiency diseases (PIDs) that cause recurrent mycobacterial infections include X-linked hyper-IgM syndrome, severe combined immunodeficiency syndrome and deficiencies of the IL-12/23-IFN-γ axis.
Sequencing of the CYBB gene documented a novel insertiondeletion mutation in exon 10 which resulted in amino acid alterations at positions 388 and 389. Over 400 different mutations of the CYBB gene have been described.6 While mutational analysis is important for definitive diagnosis, it is not routinely available, however, and immunological evaluation with neutrophil burst testing is sufficient to make a diagnosis and initiate treatment. Prophylactic antibiotic therapy with cotrimoxazole has been effective in reducing serious bacterial infections, while prophylactic itraconazole has substantially decreased the number of Aspergillus infections.1,2 Interferon-gamma has also been shown to be a highly effective prophylactic therapy, but the cost remains prohibitive in resource-limited settings.1
Improved recognition of CGD and early diagnosis is the cornerstone in reducing morbidity and mortality, so that prophylaxis may be initiated early, infections may be treated promptly and possibly curative haematopoietic stem cell transplantation considered.
Acknowledgment. Dr R Naidoo is an infectious diseases Fellow supported by PEPFAR/USAID through the ANOVA Health Institute.
References
1. Holland S. Chronic granulomatous disease. Clinic Rev Allerg Immunol 2010;38:3-10. [ Links ]
2. De Oliveira Jnr EB, Bustamante J, Newberger PE, Condino-Neto A. The human NADPH oxidase: primary and secondary defects impairing the respiratory burst function and the microbicidal ability of phagocytes. Scand J Immunol 2011;73(5):420-427. [ Links ]
3. Naidoo R, Ungerer L, Cooper M, Pienaar S, Eley BS. Primary Immunodeficiencies: A 27-year review at a tertiary paediatric hospital in Cape Town, South Africa. J Clin Immunol 2011;31:99-105. [ Links ]
4. Lee PPW, Chan KW, Jiang LP, et al. Susceptibility to mycobacterial infections in children with X-linked chronic granulomatous disease. Ped Infect Dis J 2008;27(3):224-230. [ Links ]
5. Bustamante J, Arias AA, Vogt G, et al. Germline mutations that selectively affect macrophages in kindreds with X-linked predisposition to tuberculous mycobacterial disease. Nat Immunol 2011;12(3):213-221. [ Links ]
6. Roos D, Kuhns DB, Maddalena A, et al. Hematologically important mutations: X-linked chronic granulomatous disease (third update). Blood Cells Mol Dis 2010;45(3):246-265. [ Links ]
Accepted 3 August 2011.
Corresponding author: R Naidoo (Reene.Naidoo@uct.ac.za)

