










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSAMJ: South African Medical Journal
On-line version ISSN 2078-5135
Print version ISSN 0256-9574
SAMJ, S. Afr. med. j. vol.101 n.7 Pretoria Jul. 2011
FORUM
CLINICAL IMAGES
A case of blinding abdominal pain
A K Mutyaba; E C Gardiner; S Murphy
A 12-year-old girl was admitted to hospital with a week's history of severe abdominal pain, distension and generalised weakness, and was managed for incomplete intestinal obstruction. Plain abdominal films were compatible with ileus, and ultrasound showed no ascites, no lymphadenopathy, and normal abdominal and pelvic organs. We saw her following the onset of new generalised tonic-clonic seizures. She was slightly obtunded with raised blood pressure (166/115 mmHg) and sinus tachycardia (130 bpm), apyrexial, and had mild psychomotor retardation and blindness with no light perception in either eye. Fundoscopy was normal. There was no meningism or sensorimotor deficit. Apart from a marginally low serum sodium level (131 mmol/l), the results of all biochemical and haematological investigations were normal. Inflammatory markers (c-reactive protein, erythrocyte sedimentation rate and procalcitonin) and cerebrospinal fluid assessment were normal.
A computed tomography (cT) scan of her brain showed bilateral hypodense parieto-occipital white matter changes consistent with oedema (Fig. 1). MRI scanning revealed prominent cortical and subcortical hyperintensities in the occipital and high parietal regions consistent with the radiological phenomenon of posterior reversible encephalopathy syndrome (PRES) (Fig. 2).
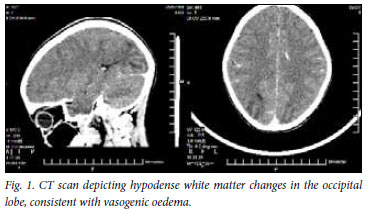
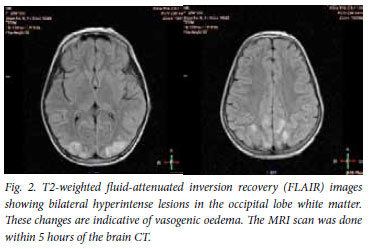
The patient's vision returned following normalisation of her blood pressure with β-blockers. A suspicion of acute intermittent porphyria (AIP) was confirmed when a markedly elevated porphyrin level was detected in her blood and urine (urinary aminolaevulanic acid 1 521 µmol/10 mmol creatinine, normal <45; urinary porphobilinogen 1 426 µmol/10 mmol creatinine, normal <16, with a positive plasma peak at 619 nm). Her abdominal pain and hyponatraemia resolved following commencement of a high-carbohydrate diet and administration of intravenous haem arginate. She was discharged 12 days after admission, completely well.
Discussion
First described by Hirchey et al. in 1996,1 PRES is a term for a radiologico-clinical entity of vasogenic cerebral oedema mainly in the parietal and occipital white matter regions, and presenting with neurological symptoms including headache, altered mental state, visual disturbances, seizures etc. PRES is seen in a variety of clinical scenarios including pre-eclampsia/eclampsia and accelerated hypertension, and less commonly in sepsis and with the use of immunosuppressive agents, e.g. tacrolimus.2 In 70 - 80% of cases, moderate to severe hypertension is observed; indeed, 'hypertensive encephalopathy' is understood to be the clinical correlate of PRES.2 Hypomagnesaemia has also been associated with PRES.3
The white matter lesions best seen on MRI represent vasogenic oedema that is reversible with removal of the underlying cause. Its predilection for the posterior cerebral region is thought to be due to a deficiency in autoregulation owing to a paucity of sympathetic innervation to the posterior vasculature.4 It is commonly accepted that a severely elevated BP exceeds the upper limits of cerebro-vascular autoregulation, causing capillary endothelial injury and subsequent leakage leading to the vasogenic oedema observed.3 An immune-mediated cascade involving T-lymphocytes, various cytokines and endothelial adhesion molecules causing endothelial dysfunction and triggering cerebral vasoconstriction and hypoperfusion, may explain the absence of a raised BP in some cases of PRES.3
We found 5 other cases of PRES occurring in AIP.4-6 Presumably, PRES observed in AIP is secondary to the accelerated hypertension that may be observed as part of the autonomic hyperactivity of AIP. Our evidence for this is in the temporal relationship between the elevated BP and onset of PRES symptomatology (blindness, seizures, psychomotor retardation) and their resolution following treatment of the hypertension. Although AIP does present with neurological symptoms (altered mental status, motor weakness, abdominal pain, autonomic hyperactivity, etc.), its pathogenetic mechanism and range of symptoms (especially its involvement of the peripheral nervous system) are quite distinct from what is observed in PRES. Furthermore, the resolution of the symptoms we attributed to PRES in the face of persisting hyperporphyrinaemia (abdominal pain persisting and urinary porphyrins still elevated after seizures and blindness resolved) makes it unlikely that they were due to the porphyrin neurotoxicity of AIP.
We thank Dr corne Rossouw of Drs Visser, Erasmus, Vawda and Partners for reporting on the MRI.
1. Hinchey J, chaves c, Appignani B, et al. A reversible posterior leukoencephalopathy syndrome. N Engl J Med 1996;334:494-500. [ Links ]
2. Bartynski WS. Posterior reversible encephalopathy syndrome, Part 1: Fundamental imaging and clinical features. Am J Neuroradiol 2008;29:1036-1042. [ Links ]
3. Bartynski WS. Posterior reversible encephalopathy syndrome, Part 2: controversies surrounding pathophysiology of vasogenic edema. Am J Neuroradiol 2008;29:1043-1049. [ Links ]
4. Utz N, Kinkel B, Hedde JP, et al. MR Imaging of acute intermittent porphyria mimicking reversible posterior leucoencephalopathy syndrome. Neuroradiology 2001;43:1059-1062. [ Links ]
5. Celik M, Forta H, Dalkilic T, et al. MRI reveals reversible lesions resembling posterior reversible encephalopathy in porphyria. Neuroradiology 2002;44:839-841. [ Links ]
6. Kang SY, Kang JH, choi Jc, et al. Posterior reversible encephalopathy syndrome in a patient with acute intermittent porphyria. J Neurol 2010;257:663-664. [ Links ]
The authors are affiliated to Livingstone Hospital, Port Elizabeth, where Arthur Mutyaba is a community service medical officer in the ICU Department; Emma Gardiner is a registrar in the Department of Internal Medicine; and Shereen Murphy is Head of Radiology Department.
Corresponding author: E Gardiner (gardiner.emmacora@gmail.com)

