










Serviços Personalizados
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkSAMJ: South African Medical Journal
versão On-line ISSN 2078-5135
versão impressa ISSN 0256-9574
SAMJ, S. Afr. med. j. vol.101 no.6 Pretoria Jun. 2011
ORIGINAL ARTICLES
Clinical characteristics and outcomes of familial and idiopathic dilated cardiomyopathy in Cape Town: a comparative study of 120 cases followed up over 14 years
Ntobeko B A NtusiI; Motasim BadriII; Freedom GumedzeIV; Ambroise WonkamV; Bongani M MayosiIII
IFCP (SA). Cardiac Clinic, Department of Medicine, Groote Schuur Hospital and University of Cape Town
IIPhD (Present address: College of Medicine, King Saud Bin Abdelaziz University of Medical Sciences, Riyadh, Kingdom of Saudi Arabia). Cardiac Clinic, Department of Medicine, Groote Schuur Hospital and University of Cape Town
IIIDPhil, FCP (SA). Cardiac Clinic, Department of Medicine, Groote Schuur Hospital and University of Cape Town
IVPhD. Department of Statistical Sciences, University of Cape Town
VMD. Division of Human Genetics, Departments of Clinical Laboratory Sciences and Medicine, Groote Schuur Hospital and University of Cape Town
ABSTRACT
BACKGROUND: It is not known whether there are differences in clinical characteristics and outcomes of patients with familial and idiopathic dilated cardiomyopathy (DCM) in an African setting.
PURPOSE: To compare the clinical characteristics and outcomes of familial and idiopathic DCM.
METHODS: We performed a retrospective study of familial and idiopathic DCM at Groote Schuur Hospital, Cape Town, between 1 February 1996 and 31 December 2009. Clinical, electrocardiographic (ECG) and imaging characteristics were compared, in addition to treatment and survival.
RESULTS: Eighty patients with idiopathic DCM and 40 familial cases were studied. ECG T-wave inversion was significantly more frequent in familial DCM (87.5%) than in idiopathic cases (68.8%) (p=0.014), whereas idiopathic patients had a higher prevalence of pathological Q waves (32.5%) than familial cases (12.5%) (p=0.028). Cardiac chambers were significantly more dilated with poorer systolic function in idiopathic than familial cases. A mortality rate of 40% after a median follow-up of 5 years was, however, similar in both groups. The presence of New York Heart Association functional class III and IV symptoms was an independent predictor of mortality (odds ratio (OR) 3.85, 95% confidence interval (CI) 1.30 - 48.47, p<0.001), while heart transplantation was an independent predictor of survival (OR 4.72, 95% CI 1.31 - 72.60, p=0.026) in both groups. Digoxin use without serum monitoring was a significant predictor of mortality in idiopathic DCM (OR 1.62, 95% CI 1.04 - 3.98, p=0.037).
CONCLUSIONS: Patients with idiopathic DCM have greater cardiac dysfunction than those with familial disease, but mortality is similarly high in both groups. Digoxin use without drug level monitoring may be associated with increased mortality in idiopathic DCM.
Cardiomyopathies are primary disorders of heart muscle associated with left ventricular dysfunction and development of heart failure, in which the heart muscle is structurally and functionally abnormal in the absence of significant coronary artery disease, hypertension, valvular disease, pericardial disease or congenital heart disease sufficient to explain the observed myocardial abnormality.1 In sub-Saharan Africa (SSA), dilated cardiomyopathy (DCM) accounts for 10 - 17% of all cardiac conditions encountered at autopsy, and for 17 -48% of hospitalisations for heart failure.2,3
The Heart of Soweto study (a contemporary study examining the characteristics and burden of cardiovascular disease in urban Africa) indicates that heart failure is the commonest cardiovascular diagnosis, with moderate to severe left ventricular dysfunction present in 53% of cases, and 68% of patients with heart failure diagnosed with DCM or hypertensive heart disease.4 While DCM occurs at any age, it is most common in the 3rd and 4th decades of life, with men affected twice as often as women. Two-thirds of patients with DCM, especially those over 55 years of age, have persistently low arterial pressure, ventricular arrhythmias and/or atrioventricular valve incompetence and die within 5 years of their first symptom.5 To the best of our knowledge, with the exception of peripartum cardiomyopathy6 there are no studies of the outcome of DCM in a contemporary African setting.
While many cases of DCM are thought to be sporadic or acquired, in up to 50% of cases the disease is inherited and is termed familial DCM.7 Familial DCM is principally caused by mutations in genes encoding cytoskeletal, nuclear and sarcomeric proteins in the cardiac myocyte, often presenting with incomplete penetrance, variable expression and significant locus and allelic heterogeneity.8 Modifying genes, lifestyle and additional factors are reported to influence onset of disease, disease progression and prognosis in familial DCM. It is of clinical interest to know whether the clinical characteristics and treatment modalities influence outcome in patients with familial DCM, and whether there are differences in clinical characteristics and outcome between those with familial and idiopathic DCM.
The purpose of this study was to compare the clinical characteristics and outcome of patients with familial and idiopathic DCM in a contemporary African setting with access to the full range of proven medical and surgical interventions for heart failure, such as cardiac resynchronisation therapy and heart transplantation, that are available at Groote Schuur Hospital (GSH) in Cape Town.9
Methods
We reviewed the medical records of all patients diagnosed with familial and idiopathic DCM and evaluated at the GSH Cardiac Clinic from 1 February 1996 to 31 December 2009. The patients were seen in a dedicated cardiomyopathy clinic, where one of the authors (BMM) has a special interest in disorders of heart muscle, and do not reflect the total experience of the GSH Cardiac Clinic. All included patients were diagnosed using established criteria for DCM, based on clinical evidence of heart failure associated with left ventricular dilatation and a left ventricular ejection fraction <50% on echocardiography or cardiac catheterisation.1 Familial DCM was defined as the finding that a patient with unexplained DCM had a first-degree relative with the condition.10 The selection of the patients for inclusion has been described elsewhere.7 The study was approved by the University of Cape Town Research Ethics Committee (REC Ref. No. 197/96).
Data collection
All patients had comprehensive clinical assessment. Information collected at the time of physical examination included history of medical co-morbidity, medications used, and history of alcohol and tobacco use. Important clinical parameters including pulse rate, blood pressure, oedema, jugular venous pressure (JVP), presence of murmurs and crepitations were recorded. Clinical assessment was complemented by chest radiography, an electrocardiogram (ECG) and detailed two-dimensional and Doppler colour-flow echocardiography. Normal values for echocardiographic measurement were based on age and body surface area. Cardiac catheterisation was performed when appropriate, and tissue obtained at biopsy of right ventricular endomyocardium was examined by light microscopy, immunohistochemistry and electron microscopy by a pathologist with no knowledge of the family history of any patient.
Statistical analysis
Results of quantitative variables are given as mean (standard deviation (SD)). Categorical variables are represented as number and percentage. Pearson's chi-square or Fisher's exact test was used to compare the relative frequency of characteristics between individuals. All p-values were two-sided, and a p-value <0.05 was considered significant.
Kaplan-Meier survival curves were constructed using the productlimit method, and were compared using the log-rank test. Kaplan-Meier curves were utilised to visualise the survival experience between familial and idiopathic DCM patients, and the focus was on mortality rather than time to death, justifying the choice of Cox's logistic regression rather than the Cox proportional hazards model. Age-, gender-and race-adjusted survival curves for the general South African population were derived and compared with the Kaplan-Meier survival rates for the patients with idiopathic or familial DCM using the chi-square test. Kaplan-Meier survival curves were constructed using STATA version 9.0 (STATA Corporation, College Station, TX, USA). Univariate and multivariate logistic regression analysis was used to determine predictors of death.
Results
Clinical characteristics at presentation
A total of 120 patients were studied, comprising 80 patients with idiopathic DCM and 40 familial cases from 29 families. The familial DCM patients had a younger age at diagnosis compared with the idiopathic DCM patients, with mean ages of 25.6 (SD 15.1) years and 39.1 (SD 12.6) years (p=0.001), respectively, as reported elsewhere.7
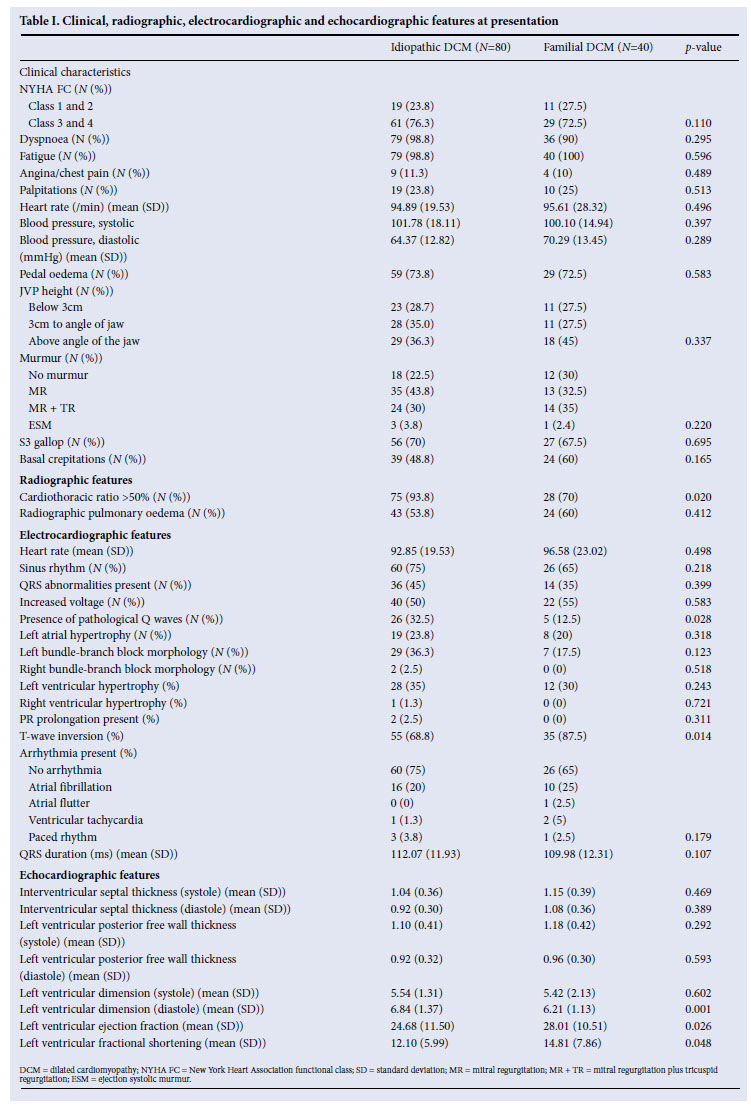
The clinical symptoms and signs of patients with familial DCM were similar to those of patients with idiopathic DCM (Table I). ECG findings were similar between the two groups, with the exception of T-wave inversion, which was found more commonly in familial patients compared with idiopathic DCM patients (35 (87.5%) v. 55 (68.8%), respectively, p=0.014), and pathological Q waves, which were found more commonly in idiopathic DCM (32.5%) than in familial cases (12.5%) (p=0.028). Patients with idiopathic DCM were more likely to have radiographic cardiomegaly, which was found in 75/80 (93.8%) of patients with idiopathic DCM compared with 28/40 (70%) of familial cases (p=0.020). The radiographic findings were corroborated by echocardiography, which showed patients with idiopathic DCM to have significantly larger left ventricular end-diastolic dimensions (LVEDD) (p=0.001), lower left ventricular ejection fraction (LVEF) (p=0.026) and fractional shortening (LVFS) (p=0.048) than the familial cases (Table I).
At cardiac catheterisation, the only difference noted was in the LVEF, which was found to be lower in patients with idiopathic DCM compared with familial cases (26.2 (SD 9.7) v. 31.07 (SD 15.63), p=0.032). There were no differences between the two groups in histological characteristics on light microscopy. There were also no significant differences between familial and idiopathic DCM patients in relation to risk factors for cardiovascular disease, lung disease, HIV infection or stroke.
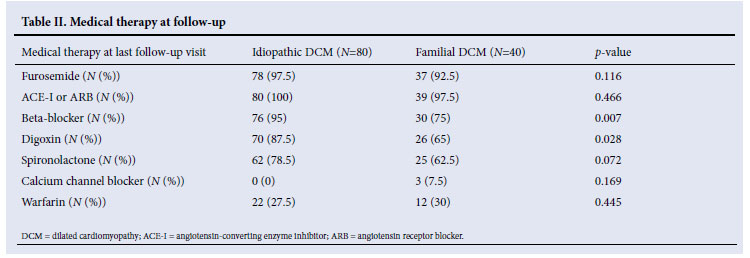
Medical therapy and outcome at last follow-up visit
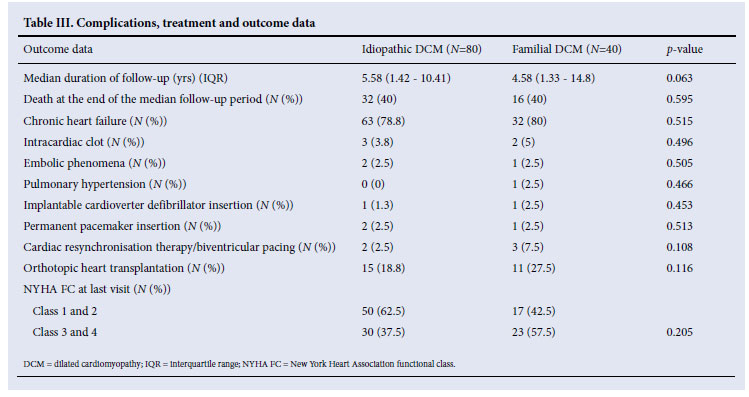
Familial and idiopathic DCM patients were on similar treatments. However, beta-blockers (p=0.007) and digoxin (p=0.028) were more commonly prescribed for patients with idiopathic DCM (Table II). There were no differences in mortality, clinical features, cardiovascular complications or utilisation of orthotopic heart transplantation between the two groups at last follow-up (Table III).
On univariate logistic regression analysis, factors that increased the likelihood of death in both familial and idiopathic cases were an increased LVEDD (p=0.038) and LVESD (p=0.049), an elevated JVP (above the angle of the jaw) at the initial visit (p=0.028), pulmonary hypertension (p=0.025), lack of orthotopic heart transplantation (p=0.004), and symptomatic heart failure with New York Heart Association (NYHA) functional class III and IV symptoms at the last visit (p=0.017), as shown in Table IVa. On multivariate analysis, significant predictors of mortality included lack of orthotopic heart transplantation (odds ratio (OR) 4.7, 95% confidence interval (CI) 1.3 -72.6, p=0.026) and NYHA class III and IV symptoms at the last visit (OR 3.8, 95% CI 1.3 -48.5, p<0.001).
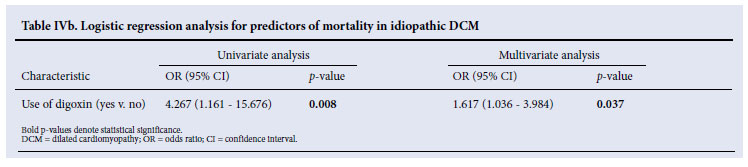
The use of digoxin in patients with idiopathic DCM was associated with increased mortality (OR 1.6, 95% CI 1.0 -3.9, p=0.037), but this was not the case in the familial DCM group (Table IVb).
Survival analysis
Fig. 1A shows the Kaplan-Meier survival analysis, with no survival difference between the idiopathic and familial DCM groups (p=0.739). The mortality rate at the end of a median follow-up period of 5 years was 40% in both groups (Table III).
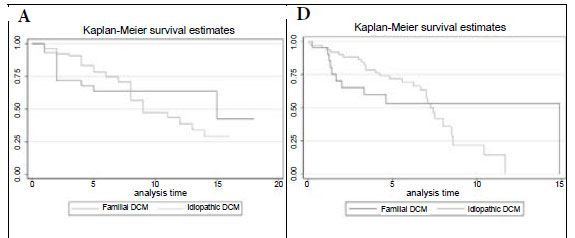
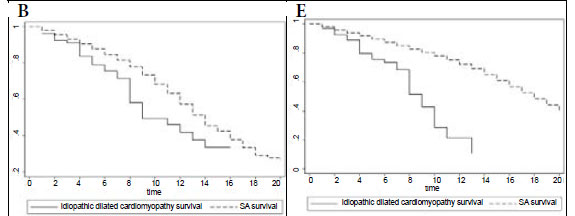
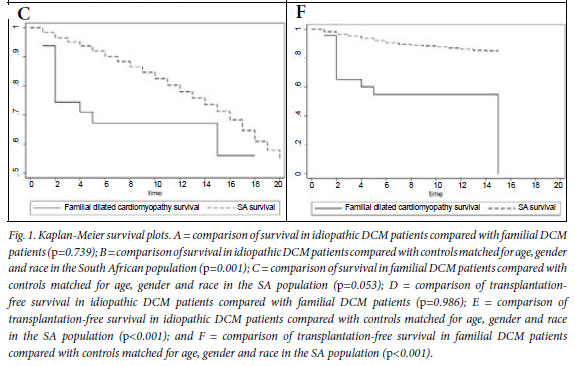
Fig. 1B compares survival of the idiopathic DCM patients with controls matched for age, gender and race in the general South African (SA) population, showing that mortality is higher in the idiopathic DCM group (p=0.001). In Fig. 1C, survival of the familial DCM cohort is compared with the matched members of the South African population, and there is a strong trend towards increased mortality in the familial DCM group (p=0.053). Fig. 1D compares transplant-free survival between idiopathic and familial DCM, showing no difference between the two groups (p=0.986). Fig. 1E compares transplant-free survival between idiopathic DCM patients and controls matched for age, gender and race in the SA population (p<0.001), and Fig. 1F compares transplant-free survival in familial DCM patients and controls matched for age, gender and race in the SA population (p<0.001). Survival free of transplantation is worse for both groups when compared with the general population.
Discussion
Several important observations emerge from this study of 80 idiopathic and 40 familial DCM patients. Firstly, despite the more favourable age and left ventricular function of the patients with familial DCM, there was a high mortality (40%) after a median follow-up of 5 years, which was similar to patients with idiopathic DCM. Second, the presence of symptoms of heart failure was the most important clinical predictor of mortality, which was significantly reduced by intervention with orthotopic heart transplantation. Thirdly, and of concern, the use of digoxin in patients with idiopathic DCM appeared to be associated with excess mortality.
We show that patients with idiopathic DCM have greater left ventricular dimension in systole and diastole on echocardiography, more radiographic cardiomegaly, and lower contractile capacity on echocardiography and cardiac catheterisation. ECGs revealed a greater prevalence of pathological Q waves in idiopathic DCM in the absence of coronary artery disease. Similarly, Grünig and colleagues have shown that ECG ST-segment and T-wave abnormalities occurred more frequently in familial than in idiopathic DCM.11 However, other authors have failed to demonstrate ECG differences between familial and idiopathic cases of DCM.12-14 ECG features are not sufficiently specific or sensitive to distinguish between familial DCM and idiopathic DCM, and are not useful for clinical classification. In our study, as with most comparisons of familial DCM with idiopathic DCM,12,15,16 there were no reliable clinical or morphological features capable of distinguishing between familial and non-familial forms of DCM, apart from a younger age of onset.7
We found that persistent symptoms of heart failure associated with NYHA functional class III or IV features are the most powerful predictor of mortality in both idiopathic and familial DCM. Similarly, Limongelli and colleagues showed that, in a cohort of 48 adolescents with idiopathic DCM, NYHA functional class III and IV, probrain natriuretic peptide and ECG left atrial enlargement were the most important predictors of adverse outcomes.17 Pasotti and co-investigators reported on a group of 27 consecutive patients with DCM and lamin A/C gene mutations and found that NYHA functional class III and IV and competitive sports were the significant predictors of all cardiovascular events, including death.18 Other important predictors of mortality in DCM patients include advanced age, protodiastolic gallop, failure of the myopathic ventricle to respond to inotropic stimulation, ventricular arrhythmias, prolonged ECG QRS duration, reduced left ventricular function, and late gadolinium enhancement on cardiovascular magnetic resonance imaging.19,20
The annual mortality rate for patients with heart failure is 10 -13%, which is similar to the mortality rate of 50% after a median followup period of 5 years that was found in this study.21 However, while the rate of progression is variable and influenced by several factors, symptomatic patients experience progressive deterioration, and 10 40% of symptomatic patients with heart failure may die within a year.22 It is therefore important that clinicians pay particular attention to medical and surgical management of persistent symptoms in DCM patients, with the aim of escalating management including use of cardiac resynchronisation therapy and heart transplantation, as NYHA class III and IV symptoms remain powerful predictors of mortality. The medical history is therefore the most important tool for assessment of adequacy of treatment in DCM.
The patients in our study were on good medical treatment, with more than 60% simultaneously receiving an angiotensin-converting enzyme inhibitor or angiotensin receptor blocker, a beta-blocker, spironolactone and digoxin. However, we found that beta-blockers and digoxin were more commonly prescribed in the idiopathic DCM group. The increased use of beta-blockers and digoxin in the idiopathic DCM group may reflect the fact that this group of patients had more severe disease, as evidenced by a greater LVEDD and lower LVEF. It has been observed by others that patients with familial DCM are less intensively treated with evidence-based therapies for heart failure than patients with sporadic DCM.16
Interestingly, digoxin appears to be a significant predictor of mortality in idiopathic DCM but not in familial DCM. Evidence from old clinical trials conducted before the introduction of betablockers for the treatment of heart failure supports the use of digoxin in patients with left ventricular systolic dysfunction for the treatment of heart failure, particularly those with advanced symptoms.23,24 There is, however, no evidence that digoxin improves survival - and in fact it may worsen outcomes in DCM patients. The effect of digoxin therapy differs between men and women, being associated with an increased risk of death from any cause among women with heart failure and depressed left ventricular systolic function.25 The DIG trial, a study of almost 6 800 patients with symptomatic heart failure and a LVEF below 45% and who were in sinus rhythm, assigned randomly to receive either digoxin or placebo, showed that after 3 years of follow-up: (i) there was no difference in survival between the digoxin and placebo groups; (ii) the patients on digoxin had reduced symptoms and hospitalisation for heart failure; (iii) patients on digoxin had significant increase in non-heart failure deaths, including deaths from arrhythmia, particularly in women; and (iv) lower serum digoxin levels correlated with survival (the ideal serum level is 0.65 - 1.0 nmol/l).26,27 Serum levels of digoxin were not monitored in our study. The results of our retrospective analysis suggest that clinicians should refrain from blanket prescription of digoxin for all DCM patients without monitoring digoxin levels.
Our findings have important implications for clinical practice and research. First, the presence of symptoms of heart failure with mild cardiac structural and functional abnormalities in relatives of patients with DCM is associated with a poor prognosis. Second, the study emphasises the need to refer patients with DCM and persistent symptoms of effort intolerance for evaluation for life-saving interventions such as heart transplantation. There is anecdotal evidence that patients with DCM are not referred in a timely manner for heart transplantation in South Africa. Finally, there is a need to review the use of digoxin in patients with heart failure in view of the mortality risk that has been shown in this study. It is prudent to monitor digoxin levels, together with serum potassium and renal function, in all patients in view of the correlation of levels with mortality.27
While this study has important findings for clinical practice, it also has several limitations. First, generalisation of findings may be limited by the relatively small sample size. Second, its population is dominated by patients with familial DCM, which is typical of a tertiary referral clinic. Finally, the study has the drawbacks imposed by a retrospective design.
In conclusion, DCM due to familial and non-familial causes is associated with a high mortality despite modern medical and surgical therapy. Patients with persistent symptoms of heart failure despite optimal medical therapy should be referred for orthotopic heart transplantation and other life-saving interventions.
We are grateful to the patients and families who participated in this study. We acknowledge the expert assistance of Ms Lebogang Montewa, Ms Stella du Toit, Mr Simpiwe Nkepu, Ms Carolina Lemmer, and Sisters Maitele Tshifularo, Unita September and Veronica Francis in the execution of this study.
We have no conflict of interest or disclaimers to declare.
The authors are funded in part by research grants from the University of Cape Town, Medical Research Council of South Africa, Discovery Foundation, the National Research Foundation of South Africa, and the Lily & Ernst Hausmann Research Trust.
References
1. Elliott P, Andersson B, Arbustini E, et al. Classification of the cardiomyopathies: a position statement from the European Society of Cardiology working group on myocardial and pericardial diseases. Eur Heart J 2008;29:270-276. [ Links ]
2. Ntusi NBA, Mayosi BM. Epidemiology of heart failure in sub-Saharan Africa. Exp Rev Cardiovasc Ther 2009;7:169-180. [ Links ]
3. Sliwa K, Damasceno A, Mayosi BM. Epidemiology and etiology of cardiomyopathy in Africa. Circulation 2005;112:3577-3583. [ Links ]
4. Sliwa K, Wilkinson D, Hansen C, et al. Spectrum of heart disease and risk factors in a black urban population in South Africa (the Heart of Soweto Study): a cohort study. Lancet 2008;371:915-922. [ Links ]
5. Freers J, Hakim J, Mayanja-Kizza H, Parry E. The heart. In: Parry E, Godfrey R, Mabey D, Gill G, eds. Principles of Medicine in Africa. 3rd ed. Cambridge, UK: Cambridge University Press, 2004:837-886. [ Links ]
6. Sliwa K, Forster O, Libhaber E, Fett JD, et al. Peripartum cardiomyopathy: inflammatory markers as predictors of outcome in 100 prospectively studied patients. Eur Heart J 2006;27(4):441-446. [ Links ]
7. Ntusi NBA, Wonkam A, Shaboodien G, Badri M, Mayosi BM. Frequency and clinical genetics of familial dilated cardiomyopathy in Cape Town: Implications for the evaluation of patients with unexplained cardiomyopathy. S Afr Med J 2011;101;xxx (this issue). TO BE COMPLETED [ Links ]
8. Moolman-Smook JC, Mayosi BM, Brink PA, Corfield VA. Molecular genetics of cardiomyopathy: changing times, shifting paradigms. Cardiovasc J S Afr 2003;14:145-155. [ Links ]
9. Mayosi BM. The UCT Department of Medicine at 90 years: Continuity and change. S Afr Med J 2010;100:548. [ Links ]
10. Mestroni L, Maisch B, McKenna WJ, et al. Guidelines for the study of familial dilated cardiomyopathies. Eur Heart J 1999;20:93-102. [ Links ]
11. Grünig E, Tasman JA, Kücherer H, Franz W, Kübler W, Katus HA. Frequency and phenotypes of familial dilated cardiomyopathy. J Am Coll Cardiol 1998;31(1):186-194. [ Links ]
12. Michels VV, Moll PP, Miller FA, et al. The frequency of familial dilated cardiomyopathy in a series of patients with idiopathic dilated cardiomyopathy. N Engl J Med 1992;326(2):77-82. [ Links ]
13. Keeling PJ, Gang Y, Smith G, et al. Familial dilated cardiomyopathy in the United Kingdom. Br Heart J 1995;73:417-421. [ Links ]
14. Zachara E, Caforio AL, Carboni GP, et al. Familial aggregation of idiopathic dilated cardiomyopathy: clinical features and pedigree analysis in 14 families. Br Heart J 1993;69(2):129-135. [ Links ]
15. Michels VV, Driscoll DJ, Miller FA, et al. Progression of familial and non-familial dilated cardiomyopathy: long term follow up. Heart 2003;89(7):757-761. [ Links ]
16. Moretti M, Merlo M, Barbati G, et al. Prognostic impact of familial screening in dilated cardiomyopathy. Eur J Heart Fail 2010;12(9):922-927. [ Links ]
17. Limongelli G, Pacileo G, Ancona R, et al. Clinical course and risk profile in adolescents with idiopathic dilated cardiomyopathy. Am J Cardiol 2010;105(5):716-720. [ Links ]
18. Pasotti M, Klersy C, Pilotto A, et al. Long-term outcome and risk stratification in dilated cardiolaminopathies. J Am Coll Cardiol 2008;52(15):1250-1260. [ Links ]
19. Hombach V, Merkle N, Torzewski J, et al. Electrocardiographic and cardiac magnetic resonance imaging parameters as predictors of a worse outcome in patients with idiopathic dilated cardiomyopathy. Eur Heart J 2009;30(16):2011-2018. [ Links ]
20. Amiya E, Tanabe K, Ikari Y, Nakajima Y, Hara K. Prolonged QRS duration and severity of mitral regurgitation are unfavorable prognostic markers of heart failure in patients with nonischemic dilated cardiomyopathy. Circ J 2006;70(1):57-62. [ Links ]
21. Konstam MA. Progress in heart failure management?: Lessons from the real world. Circulation 2000;102(10):1076-1078. [ Links ]
22. Deedwania PC. The key to unraveling the mystery of mortality in heart failure: An integrated approach. Circulation 2003;107(13):1719-1721. [ Links ]
23. Lee DC-S, Johnson RA, Bingham JB, et al. Heart failure in outpatients. N Engl J Med 1982;306(12):699-705. [ Links ]
24. Group TC-DMR, Cohn J, Hawkins M, et al. Comparative effects of therapy with captopril and digoxin in patients with mild to moderate heart failure. JAMA 1988;259:539-544. [ Links ]
25. Rathore SS, Wang Y, Krumholz HM. Sex-based differences in the effect of digoxin for the treatment of heart failure. N Engl J Med 2002;347(18):1403-1411. [ Links ]
26. The effect of digoxin on mortality and morbidity in patients with heart failure. The Digitalis Investigation Group. N Engl J Med 1997;336:525-533. [ Links ]
27. Rathore SS, Curtis JP, Wang Y, Bristow MR, Krumholz HM. Association of serum digoxin concentration and outcomes in patients with heart failure. JAMA 2003;289:871-878. [ Links ]
Accepted 21 March 2011.
Corresponding author: B M Mayosi (bongani.mayosi@uct.ac.za)


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}