










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSAMJ: South African Medical Journal
On-line version ISSN 2078-5135
Print version ISSN 0256-9574
SAMJ, S. Afr. med. j. vol.101 n.3 Pretoria Mar. 2011
ORIGINAL ARTICLES
Phaeochromocytoma in black South Africans - a 30-year audit
K R L Huddle
MB BCh, FCP (SA), FRCP. Department of Internal Medicine, Chris Hani Baragwanath Hospital, Faculty of Health Sciences, University of the Witwatersrand, Johannesburg
ABSTRACT
OBJECTIVE: Phaeochromocytomas are catecholamine-secreting tumours, the majority of which arise from the adrenal medulla. Untreated, they are potentially lethal; early diagnosis and treatment offer a good chance of cure. They are rarely reported in blacks. The clinical presentation and outcome of phaeochromocytoma in a large cohort of black South Africans is reviewed.
METHODS: Patients' records in a tertiary care university hospital were reviewed. Fifty-four black patients presenting with phaeochromocytoma between 1980 and 2009 were included. The clinical presenting features, tumour localisation and outcome were assessed.
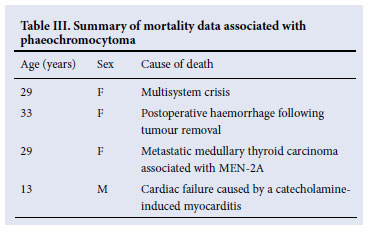
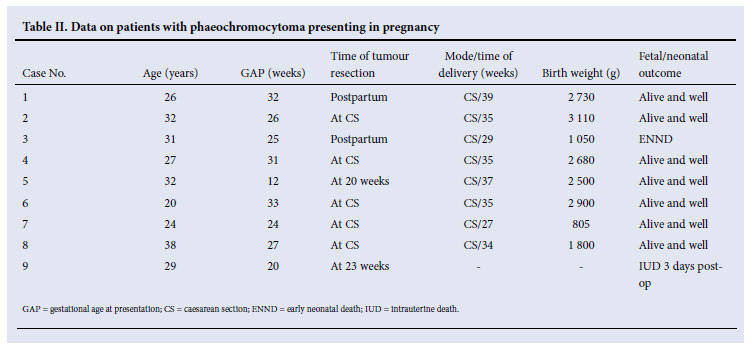
RESULTS: Fifty-four (41 female, 13 male; age range 8 - 57 years) patients were identified. Five (9%) had familial syndromes; 49 (91%) were deemed sporadic. All tumours were intra-abdominal: 34 (61%) were adrenal and 22 (39%) extra-adrenal in origin. The most common symptoms were headache (77%), palpitations (77%), and sweating (74%). All were hypertensive, almost equally divided between paroxysmal and sustained hypertension. Six (11%) presented in congestive cardiac failure including 2 with catecholamine-induced myocarditis. Two patients had features which simulated hypertrophic obstructive cardiomyopathy. Nine women presented in pregnancy: there was no maternal mortality; fetal mortality included 1 early neonatal death and 1 intrauterine death. There were 4 deaths: 1 from postoperative haemorrhage, 1 from multisystem crisis, 1 from metastatic medullary thyroid carcinoma, and 1 from catecholamine-induced myocarditis.
CONCLUSION: Phaeochromocytoma is an important although rare tumour in blacks, with similar clinical presentations and complications to those in white patients. Timely diagnosis and appropriate treatment resulted in a favourable outcome in over 90% of patients in this study.
Phaeochromocytomas are catecholamine-secreting tumours of neuroectodermal origin. Approximately 85% arise from chromaffin cells in the adrenal medulla, and 15% arise from chromaffin tissue in extra-adrenal sites extending from the neck to the pelvis, although most are found intra-abdominally. Extra-adrenal phaeochromocytomas are also referred to as paragangliomas.
The prevalence of phaeochromocytoma in patients with hypertension is estimated to be 0.1 -0.6%.1 Although these tumours are rare, their detection is of the utmost importance - they are potentially lethal owing, firstly, to their ability to secrete catecholamines, often with catastrophic consequences, and, secondly, to their potential to become malignant. The frequency of malignant phaeochromocytomas ranges from 3% to 36%.2 Early detection and removal offers a real chance of cure.
Although most phaeochromocytomas are sporadic, up to 24% of patients with apparently sporadic phaeochromocytoma may be carriers of mutations.3 The familial syndromes associated with phaeochromocytoma are inherited in an autosomal dominant manner and include multiple endocrine neoplasia type 2 (MEN-2), von Hippel-Lindau (VHL) syndrome, neurofibromatosis type 1 (NF-1), and familial paraganglioma. Germline mutations for each of these syndromes have been identified and allow screening of high-risk individuals.1
The clinical presentation of phaeochromocytoma varies, and the diagnosis therefore requires a high index of suspicion. If suspected clinically, the presence of phaeochromocytoma should first be confirmed biochemically, followed by tumour localisation. Tumour removal (if feasible) is performed after stabilisation using an α-adrenergic blocker.
Phaeochromocytomas occur in all races, but have been predominantly reported in Caucasians.2 Reports of this tumour from sub-Saharan countries are rare, consisting of single case studies or small series.4,5 In this study, the clinical presentation, tumour localisation, and outcome of phaeochromocytoma in black patients presenting to a tertiary care centre were determined. It is the largest reported from sub-Saharan Africa and is an extension of an earlier report.6
Patients and methods
Chris Hani Baragwanath Hospital (CHBH) is a 3 000-bed University of the Witwatersrand tertiary referral teaching hospital. It serves the historically disadvantaged community of Soweto and beyond, numbering several million people, almost all of whom are black.
Hospital records of patients presenting with phaeochromocytoma to the Endocrine Unit of CHBH between 1980 and 2009 were reviewed. All patients were seen and managed by the author. In all cases (except the patient diagnosed at autopsy), the diagnosis of phaeochromocytoma was confirmed biochemically prior to surgery and confirmed histopathologically. This study was approved unconditionally by the Human Research Ethics Committee (Medical) of the University of the Witwatersrand - Clearance certificate: M090960.
Results
Fifty-four patients with phaeochromocytoma were seen between 1980 and 2009. Their clinical characteristics are shown in Table I. All were black: 51 from South Africa, 2 from Botswana and 1 from the Democratic Republic of Congo; 41 were female and 13 male (F:M, 3.2:1). Ages ranged between 8 and 57 years, and 8 were under 16 years old (Fig. 1). All diagnoses of phaeochromocytoma were made during life, with one exception, who was diagnosed at autopsy following a presentation suggestive of a phaeochromocytoma-induced multisystem crisis: shock, disseminated intravascular coagulation, renal failure, and metabolic acidosis. An extra-adrenal tumour was found at autopsy. Forty-nine patients (91%) were classified as sporadic, and 5 (9%) were associated with familial syndromes. Nine tumours were detected during pregnancy. Malignancy was diagnosed in 3 patients.
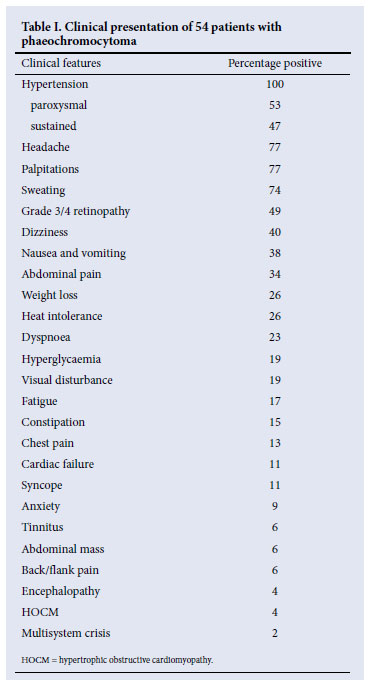
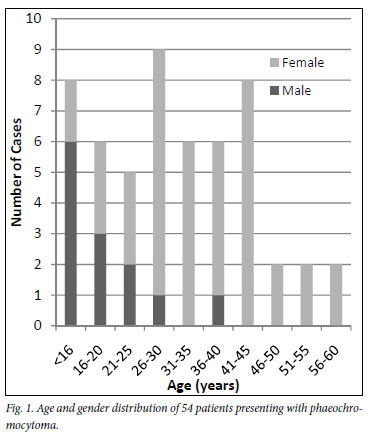
All tumours were intra-abdominal: 34 (61%) were adrenal in origin (2 patients had bilateral adrenal tumours); 22 (39%) were extra-adrenal including 2 in the Organ of Zuckerkandl and 1 in the urinary bladder. Tumour size was recorded in 49 cases with benign tumours and ranged from 3 cm × 3 cm × 3 cm to the largest at 19 cm × 16 cm × 17 cm (Fig. 2), with an average size of 6.9 cm × 5.6 cm × 4.2 cm.
Selected aspects
Cardiovascular complications
Hypertension (blood pressure >140/90 mmHg) was present in 100% of patients. In approximately half of them, the hypertension was classified as paroxysmal and was sustained in the other half. This distinction was not always easy as the blood pressure was often labile, even in patients with sustained hypertension. As indices of the severity of hypertension, 49% of patients had evidence of grades 3 or 4 hypertensive retinopathy, 2 patients presented with hypertensive encephalopathy, and 82% had clinical, electrocardiographic or echocardiographic evidence of left ventricular hypertrophy. Only 1 patient had established renal failure which was secondary to multisystem crisis, although patients often presented with an isolated elevation of the serum urea which subsequently normalised on α-adrenergic blockade and volume repletion.
Six (11%) patients presented in congestive cardiac failure. An 8-year-old girl (previously reported)7 presented in cardiogenic shock. It took 2 months on medical therapy to stabilise her cardiac function to allow the removal of an adrenal phaeochromocytoma, and a further 4 months for full recovery of cardiac function. A 16-year-old boy developed irreversible pulmonary oedema despite comprehensive medical therapy while awaiting surgery. At autopsy, the heart showed left ventricular hypertrophy with widespread myocytolysis and an inflammatory cell infiltrate - findings in keeping with a catecholamineinduced cardiomyopathy. In the remaining 4 patients, heart failure was attributed to hypertensive heart disease which responded rapidly to medical therapy, including α-adrenergic blockade. Two patients (previously reported)8 presented with clinical and echocardiographic features simulating hypertrophic obstructive cardiomyopathy. Both had successful tumour removal that led to regression of the abnormal clinical features, normalisation of the electrocardiographs, but only partial regression of the echocardiographic features, after follow-up for 24 and 32 months respectively.
Pregnancy
Nine patients presented during pregnancy (Table II), the first 4 of whom were reported previously.9 Seven women presented in the third, 1 in the second, and 1 at the end of the first trimester. All patients, with the exception of Case 8, were delivered by caesarean section (CS). Tumour resection took place at the time of CS in 5 patients, and several weeks postpartum in 2 patients. Case 5 had a successful tumour removal at 20 weeks' gestation followed 17 weeks later by CS. Case 9 had tumour resection at 23 weeks. There was no maternal mortality. In Case 3, signs of fetal distress appeared at 29 weeks' gestation that necessitated emergency CS. The premature baby had poor Apgar scores and died 3 days later from respiratory distress syndrome and intraventricular haemorrhage. Case 9 had bilateral adrenal phaeochromocytomas removed at 23 weeks, with the intention of continuing medical therapy until fetal maturity was attained. However, she experienced an intra-uterine death 3 days postoperatively while on medical therapy.
Familial syndromes
Familial syndromes were diagnosed in 5 (9%) patients. Two had typical clinical features of NF-1. One patient (previously reported)10 was diagnosed with MEN-2A based on the presence of metastatic medullary carcinoma of the thyroid associated with an adrenal phaeochromocytoma. A missense mutation in codon 634 of exon 11 of the RET gene was found. Her phaeochromocytoma was successfully removed but she refused further treatment for her thyroid malignancy which had spread to the liver. She died 8 years later. Three family members were shown to be carrying the mutant RET gene, although they showed no clinical abnormalities. They declined further investigation.
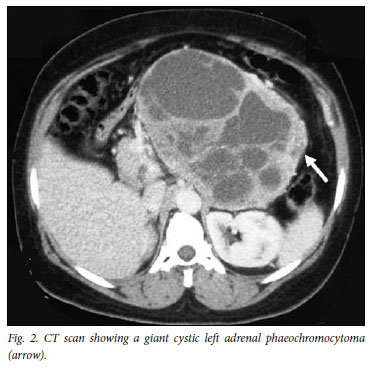
Two patients had VHL syndrome (submitted for publication). In the first, the diagnosis was based on the presence of a symptomatic cerebellar haemangioblastoma (Fig. 3) in association with a mildly symptomatic adrenal phaeochromocytoma. Genetic testing revealed a missense mutation (c.256C>T) in the VHL gene on chromosome 3p. He underwent successful removal of the phaeochromocytoma followed by removal of the cerebellar haemangioblastoma. It subsequently became apparent that his father had had a brain tumour removed many years previously. Further investigation revealed bilateral renal cell carcinoma for which he received treatment. The father and his daughter screened positive for the VHL gene. Although the daughter is clinically well, she has evidence on MRI of haemangioblastomas in her brain and spinal cord. All 3 affected members of this family are being regularly monitored for the development of complications associated with the VHL syndrome. The second diagnosis of VHL syndrome was made in the pregnant woman (Case 9) who had bilateral adrenal phaeochromocytomas. There were no clinical features of VHL syndrome; however, MRI showed a haemangioblastoma in the cervical spine. Genetic testing revealed a missense mutation (c.499C>T) in the VHL gene, located on chromosome 3p26-p25. Her 6-year-old daughter screened positive for the mutation; her 8-year-old son was negative. Both mother and daughter will be investigated further and monitored long-term.
Malignancy
Two children had evidence of malignant disease. The 8-year-old girl who had originally presented in cardiogenic shock owing to a catecholamine-induced cardiomyopathy (see above), presented 5 years later with a second adrenal phaeochromocytoma with metastatic spread surrounding the superior mesenteric artery. The adrenal tumour was removed but the metastasis was deemed unresectable. She received a therapeutic dose of 131I-metaiodobenzylguanidine (MIBG) but has biochemical and radiological evidence of residual tumour. Her blood pressure is controlled with an α-adrenergic blocker combined with a calcium channel blocker. She is relatively well and attends school, 10 years after the initial presentation.
An 11-year-old boy was diagnosed with a massive abdominal phaeochromocytoma which was only amenable to debulking. MIBG scan revealed local spread and a metastasis to the skull. He subsequently received 2 therapeutic doses of MIBG. He is relatively well 8 years after presentation, requiring α- and β-adrenergic blockers plus a calcium channel blocker to control his blood pressure.
An adult of 44 years had invasion of an abdominal lymph node by tumour following resection of an extra-adrenal phaeochromocytoma. The postoperative urinary metanephrines and MIBG scan were normal. Close follow-up has been implemented.
Vesical phaeochromocytoma
A 21-year-old man with a history of hypertension complained of repeated episodes of dizziness, palpitations and headache immediately following micturition.11 This was associated with a feeling of impending doom. A phaeochromocytoma was detected in the bladder wall and successfully resected.
Outcomes
A summary of mortality data associated with phaeochromocytoma in this study is shown in Table III. The 2 fetal/neonatal losses are shown in Table II.
Discussion
This study indicates that phaeochromocytoma is an important although rare tumour in black patients, with similar clinical presentations and complications to those in other reports.2,12,13 The paucity of reports from sub-Saharan Africa most likely reflects limited health care resources in these countries in general, and limited laboratory and radiological facilities for diagnosis of phaeochromocytoma in particular. Clearly, because of its rarity, it is not cost-effective to screen all hypertensive patients for this tumour. Rather, one should investigate those patients in whom there is a high index of suspicion based on clinical presentation:14 patients with episodic symptoms of headache, palpitations and sweating; unexplained paroxysms of arrhythmias and/or hypertension during intubation, induction of anaesthesia, tumour manipulation, and parturition; adverse cardiovascular responses to ingestion, inhalation, or injection of certain drugs; spells or attacks occurring during exertion, twisting and turning of the torso, straining, coitus or micturition; family history of phaeochromocytoma or familial syndrome; and incidental adrenal/abdominal masses. Approximately 90% of patients with phaeochromocytoma are hypertensive, with similar proportions of patients with paroxysmal and sustained hypertension.2,13,14 Postural hypotension may occur in up to 12% of patients. In the present study, headache (77%), palpitations (77%) and sweating (74%) were the most common symptoms. This triad of symptoms presenting together in a hypertensive patient has been reported as having a sensitivity of 90.9% and a specificity of 93.8%.14 All the patients in this report were hypertensive, almost equally divided between paroxysmal and sustained hypertension. Dizziness (40%) was also a common symptom; 6 (11%) patients had syncopal episodes which, on the basis of circumstantial evidence, were attributed to postural hypotension.
A cause for concern is that the diagnosis of phaeochromocytoma is often missed. For example, in a series from the Mayo Clinic, 41 of 54 autopsy-proven cases were unsuspected clinically during life.15 The many reasons for non-diagnosis include the nonspecific nature of the symptoms, the clinicians' low index of suspicion, and that some tumours are asymptomatic because they are non-functioning or secrete relatively small amounts of catecholamines.13 Our hospital has no reliable autopsy data to assess the situation, but it is highly probable that phaeochromocytomas are missed. The patient presenting with multisystem crisis was only diagnosed at autopsy.
Cardiac complications of phaeochromocytoma are many and varied, including left ventricular hypertrophy (usually concentric), catecholamine-induced myocarditis and dilated cardiomyopathy, takotsubo cardiomyopathy, and a condition simulating hypertrophic obstructive cardiomyopathy (HOCM).2 Catecholamine-induced myocarditis may result in cardiac failure and death, often without manifesting hypertension.16 Of the 6 patients in this study who presented in heart failure, 2 were assessed as having catecholamineinduced myocarditis; one showed full recovery of cardiac function several months after tumour removal; the other had a fatal outcome prior to tumour resection. Unexplained cardiac failure/pulmonary oedema should make one consider phaeochromocytoma in the differential diagnosis. Left ventricular hypertrophy associated with phaeochromocytoma may rarely be asymmetrical and mimic the features of HOCM, as was the case in 2 of our patients, showing reversibility several years after tumour removal.
Phaeochromocytoma in pregnancy may have devastating consequences for both mother and fetus. If undiagnosed during pregnancy, phaeochromocytoma is associated with a maternal and fetal mortality of around 50%.17 When the diagnosis of phaeochromocytoma is made antepartum and when appropriate treatment is instituted, maternal mortality has fallen to as low as 2%, and fetal mortality to 11%.18 The main differential diagnosis in pregnancy is pre-eclampsia, which is far more common. Pre-eclampsia occurs in the second half of pregnancy and is not associated with the typical symptoms associated with phaeochromocytoma. Pregnant women with severe/paroxysmal hypertension, symptoms suggestive of phaeochromocytoma, abnormal glucose tolerance, and sudden collapse should be investigated for phaeochromocytoma. In this study there was no maternal mortality. There was 1 early neonatal death at 29 weeks and 1 intra-uterine death at 23 weeks' gestation. Gratifyingly, the remaining 7 babies made good progress.
Significant advances have been made in determining the genetic basis of phaeochromocytomas; they are associated with the following familial syndromes: MEN-2 (activating germline mutations in the RET proto-oncogene), VHL syndrome (germline mutations in the VHL tumour suppressor gene), NF-1 (germline mutations in the NF-1 tumour suppressor gene), and phaeochromocytoma/ paraganglioma syndromes (germline mutations in the B and D subunits of mitochondrial succinate dehydrogenase - SDHB and SDHD). Genetic testing should be considered in the following situations: a positive family history, clinical features of syndromic disorder (e.g. MEN-2, VHL), children and young adults with phaeochromocytoma, bilateral adrenal tumours or multifocal extra-adrenal disease, and paragangliomas. The clinical picture and biochemical phenotype can direct the specific genetic testing required.1 There are significant benefits associated with genetic testing including the early diagnosis of other tumours in a patient with a syndromic disorder (e.g. renal cell carcinoma in VHL), allowing earlier treatment and improved prognosis; affected family members would likewise benefit. In addition, close follow-up of patients with germline mutations is indicated to detect additional chromaffin tumours or recurrences. Our 5 patients identified with familial syndromes are probably an underestimate of the number of patients with genetic disorders, as selective genetic testing has only recently become available to us.
Conclusion
Phaeochromocytomas will continue to fascinate, confound and humble. Their detection via good sleuth work is intellectually very satisfying and, in most cases, results in their cure. This report highlights the fact that phaeochromocytoma is an important although rare tumour in blacks, has a similar profile to that found in whites, and that with timely intervention a good outcome can be achieved in most patients. Future advances in the understanding of this tumour will lead to improved detection, more effective treatment - especially for malignant disease - and better outcomes.
I gratefully acknowledge the valuable assistance given by members of the following departments/divisions regarding referral of patients, their investigation and treatment: Endocrinology, Surgery, Obstetrics, Paediatrics, Radiology and Nuclear Medicine, Anatomical and Chemical Pathology, and Human Genetics of CHBH, Charlotte Maxeke Johannesburg Academic Hospital, and the National Health Laboratory Services.
References
1. Lenders JWM, Eisenhofer G, Mannelli M, Pacak K. Phaeochromocytoma. Lancet 2005;366:665-675. [ Links ]
2. Pacak K, Lenders JWM, Eisenhofer G. Pheochromocytoma: diagnosis, localization and treatment. Malden MA: Blackwell Publishing, 2007. [ Links ]
3. Neumann HPH, Bausch B, McWhinney SR, et al. Germ-line mutations in nonsyndromic pheochromocytoma. N Engl J Med 2002;346:1459-1466. [ Links ]
4. Sidibe EH. Phaeochromocytoma in Africa: rarity, gravity, and ectopy. Ann Urol 2001;35:17-21 (French). [ Links ]
5. Johnson O. Phaeochromocytoma: experience with 12 cases in Tikur Anbessa, Addis Ababa, Ethiopia. East and Central African Journal of Surgery 2004;9:71-75. [ Links ]
6. Huddle KRL, Mannell A, James MFM, Plant ME. Phaeochromocytoma. A report of 10 patients. S Afr Med J 1991;79:217-220. [ Links ]
7. Rennert W, Karbanee I, Huddle K. Phaeochromocytoma presenting as cardiogenic shock in an 8-yearold child. The Journal of Endocrinology, Metabolism and Diabetes of South Africa 2002;7:13-14. [ Links ]
8. Huddle KR, Kalliatakis B, Skoularigis J. Pheochromocytoma associated with clinical and echocardiographic features simulating hypertrophic obstructive cardiomyopathy. Chest 1996;109:13941397. [ Links ]
9. Huddle KRL, Nagar A. Phaeochromocytoma in pregnancy. Aust NZ J Obstet Gynaecol 1999;39:203-206. [ Links ]
10. Hopley M, Huddle KRL. Multiple endocrine neoplasia type 2A in a black South African family. S Afr Med J 1997;87:371-372. [ Links ]
11. Huddle KRL. Phaeochromocytoma by way of case reports. Cardiovasc J S Afr 2002;13:205-208. [ Links ]
12. Ross EJ, Griffith DNW. The clinical presentation of phaeochromocytoma. Q J Med 1989;71:485-496. [ Links ]
13. Manger WM, Gifford RW. Clinical and experimental pheochromocytoma. Cambridge MA: Blackwell Science, 1996. [ Links ]
14. Bravo EL, Tagle R. Pheochromocytoma : state-of-the-art and future prospects. Endocrine Reviews 2003;24:539-553. [ Links ]
15. Sutton MG, Sheps SG, Lie JT. Prevalence of clinically unsuspected pheochromocytoma. Review of 50year autopsy series. Mayo Clin Proc 1981;56:354-360. [ Links ]
16. Sardesai SH, Mourant AJ, Sivathondon Y, Farrow R, Gibbons DO. Phaeochromocytoma and catecholamine induced cardiomyopathy presenting as heart failure. Br Heart J 1990;63:234-237. [ Links ]
17. Schenker JG, Granat M. Phaeochromocytoma and pregnancy - an updated appraisal. Aust NZJ Obstet Gynaec 1982;22:1-10. [ Links ]
18. Ahlawat SK, Jain S, Kumari S, Varma S, Sharma BK. Pheochromocytoma associated with pregnancy: case report and review of the literature. Obstet Gynaecol Surv 1999;54:728-737. [ Links ]
Accepted 24 June 2010.
Corresponding author: K Huddle (kenneth.huddle@wits.ac.za)


{kind=link}