










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSAMJ: South African Medical Journal
On-line version ISSN 2078-5135
Print version ISSN 0256-9574
SAMJ, S. Afr. med. j. vol.100 n.6 Pretoria Jun. 2010
GUIDELINE
Guideline: appropriate use of tigecycline
A J Brink; D Bizos; K D Boffard; C Feldman; D C Grolman; J Pretorius; G A Richards; M Senekal; E Steyn; N Welkovic
ABSTRACT
INTRODUCTION: Tigecycline, the first of a new class of antibiotics, the glycylcyclines, was licensed in South Africa for the parenteral treatment of adult patients with complicated intra-abdominal infections (cIAIs) and complicated skin and soft-tissue infections (cSSTIs).
METHODS: A multidisciplinary meeting representative of the Association of Surgeons of South Africa, the Critical Care Society of Southern Africa, the Federation of Infectious Diseases Societies of Southern Africa, the South African Thoracic Society and the Trauma Society of South Africa was held to draw up a national guideline for the appropriate use of tigecycline. Background information reviewed included randomised controlled trials, other relevant publications and local antibiotic susceptibility patterns. The initial document was drafted at the meeting. Subsequent drafts were circulated to members of the working group for modification.
OUTPUT: The guideline addresses several important aspects of the new agent, summarising key clinical data and highlighting important considerations with the use of the drug. The recommendations in this guideline are based on currently available scientific evidence together with the consensus opinion of the authors.
CONCLUSION: This statement was written out of concern regarding the widespread misuse of antibiotics. Its primary intention is to facilitate heterogeneous use of antibiotics as a component of antibiotic stewardship and to highlight
1. Introduction
Tigecycline, the first of a new class of broad-spectrum antibiotics, the glycylcyclines, was recently licensed in South Africa for the parenteral treatment of adult patients with complicated intra-abdominal infections (cIAIs) and complicated skin and soft-tissue infections (cSSTIs). This statement addresses important aspects of the new agent, including pharmacokinetics, mode of action and antibacterial spectrum, summarises key clinical trial data, and highlights appropriate use of the drug. Several other important considerations are also briefly addressed.
2. Metabolism and pharmacokinetics
• Tigecycline undergoes minimal metabolism and is primarily excreted by the liver. Additional routes of elimination include renal excretion (22%).1,2
• Dosing (100 mg loading dose followed by 50 mg 12-hourly) is uncomplicated as the agent has no effect on cytochrome P450, has no clinically relevant drug interactions, the pharmacokinetics (PK) are not influenced by renal impairment, and it is not removed by haemodialysis.2
• In patients with severe hepatic impairment (Child Pugh C), the initial dose of tigecycline should be 100 mg followed by a reduced maintenance dose of 25 mg every 12 hours.
• Tigecycline protein binding is between 71% and 89% and the drug demonstrates unusual pharmacokinetics. Following multiple doses of 30-minute infusions of 50 mg given 12-hourly, the following has been documented3 (comparative PK data with 100 mg are shown in Table I):4
• a long terminal half-life (t½) of 42 hours
• a maximum peak plasma concentration (Cmax ) of 0.87 mg/l
• a minimum trough plasma concentration (Cmin ) of 0.13 mg/l
• an AUC from 0 to 24 hours (AUC0-24) of 4.7 mg/h/ml
• a high average steady-state volume of distribution (VSS) of 639 l.
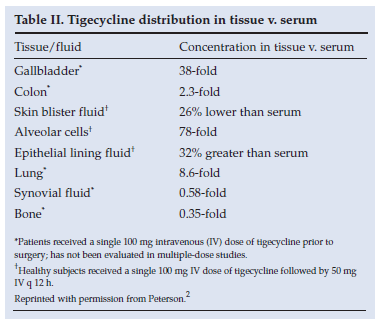
This indicates that the drug is widely distributed and undergoes extensive and rapid transfer from the blood into the tissues, where levels far exceed those of serum (Table II2).
3. Mode of action
• Tigecycline binds to the 30S ribosomal subunit, inhibiting protein synthesis in a fashion similar to that of the aminoglycosides, macrolides and linezolid.
• It is generally bacteriostatic except for Streptococcus pneumoniae and Legionella spp. where it is bactericidal. Tigecycline has a prolonged post-antibiotic effect, ranging from 4.9 hours for Escherichia coli to 3.4 - 4 hours for Staphylococcus aureus. 5 This suggests that it has the potential to exert a significant antibacterial effect even when levels are below the minimum inhibitory concentration (MIC).
• Pharmacodynamic studies have shown that tigecycline exhibits time-dependent killing, and as such it has been recommended that levels should be maintained above the MIC for 50 - 75% of the dosing interval.6 However, because of the long half-life and post-antibiotic effect, the area under the inhibitory curve (AUC/MIC) has also been shown to be most predictive of efficacy.6
• Tigecycline appears to overcome the major mechanisms conferring resistance to the tetracyclines (ribosomal protection and efflux pumps) owing to the steric hindrance afforded by a large D-ring substitution.1
• It is also not affected by common resistance mechanisms that affect other antibiotics, such as penicillin-binding protein modifications by methicillin-resistant S. aureus (MRSA), extended-spectrum β-lactamase (ESBL) production by Enterobacteriaceae, carbapenemase production by Klebsiella pneumoniae, or DNA gyrase mutations that confer fluoroquinolone resistance.2
4. Antibacterial spectrum
• The Tigecycline Evaluation and Surveillance Trial (TEST), a global, multicentre surveillance programme, documented that tigecycline is highly active against Gram-positive pathogens, including MRSA, methicillin-resistant S. epidermidis (MRSE) and enterococci, including vancomycinresistant enterococci (VRE).7,8
• The agent is also highly active against Enterobacteriaceae, anaerobes and atypical pathogens.1,2,7,8
• Reduced activity of tigecycline has been observed for Proteus spp., Providencia spp. and Morganella spp., and Pseudomonas aeruginosa is also not reliably inhibited by tigecycline.1,2
• There is neither synergy nor antagonism with other antibiotics.9
5. Clinical trial data
5.1 Tigecycline has been studied in four phase III double-blind, randomised, multicentre comparator clinical studies in adult hospitalised patients with cIAI and cSSTI:
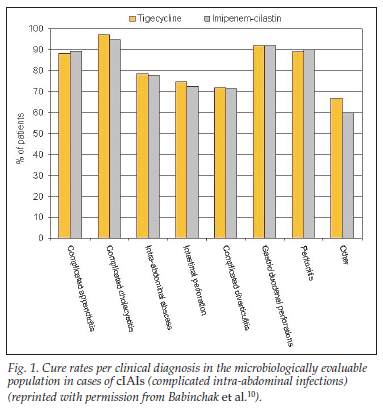
• Tigecycline, in conjunction with operative intervention, was shown to be as effective as imipenem/cilastatin in treating cIAI, with cure rates in the microbiologically evaluable patients identical in the two groups (86%, N=685 v. 86%, N=679, respectively), and the outcome did not differ between monomicrobial versus polymicrobial infections.10
• Cure rates by clinical diagnosis in the microbiologically evaluable population in cIAI are depicted in Fig. 1.
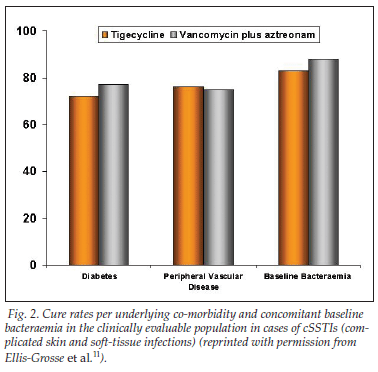
• Cure rates in clinically evaluable patients hospitalised with cSSTI were also similar for those treated with tigecycline and those treated with vancomycin plus aztreonam (87%, N=422 v. 89%, N=411, respectively).11
• Cure rates according to underlying co-morbidity and concomitant baseline bacteraemia in the clinically evaluable population in cases of cSSTI are depicted in Fig. 2.
5.2 Tigecycline was also evaluated in adults hospitalised with community-acquired pneumonia (CAP) in two randomised, double-blind, active-controlled, multinational, multicentre studies12 with levofloxacin (500 mg IVI once or twice daily) as comparator:
• Microbiological and clinical outcome was similar in the two studies, including cases with L. pneumophila infections.
• An analysis of patients with a higher risk of mortality (age >50 years, pneumonia severity index (PSI) score >3 or S. pneumoniae bacteraemia) in these two studies also showed similar results for clinically evaluable patients; favourable outcomes were documented in 89.55% (188/210) and 81.2% (152/187) in the tigecycline and levofloxacin arms, respectively.
5.3 The agent was also studied in a phase III, open-label, noncomparative study of the treatment of serious infections due to resistant Gram-negative organisms.13 In the microbiologically evaluable (ME) population at test of cure (TOC), the clinical cure rate was 72.2% (95% confidence interval (CI) 54.8 - 85.8) and the microbiological eradication rate was 66.7% (95% CI 13.7 - 78.8).
5.4 The efficacy of tigecycline compared with vancomycin or linezolid for the treatment of serious infections with MRSA or VRE was studied in another phase III, multicentre, doubleblind, randomised study.14 For MRSA infections, clinical cure rates in the ME patients (N=117) were 81.4% with tigecycline and 83.9% (N=31) with vancomycin. In patients with VRE (N=15), 3 of 3 ME patients were cured by tigecycline compared with 2 of 3 patients by linezolid.
6. Safety and tolerability
• The most frequent adverse events in all study subjects, including tigecycline-treated patients and patients receiving comparator therapy, were nausea (30% and 16%, respectively) and vomiting (20% and 11%, respectively).10,11
• No significant difference in discontinuation due to treatment-related adverse events was noted between tigecycline and all comparators (5% and 4.7%, respectively).10,11
7. Other considerations
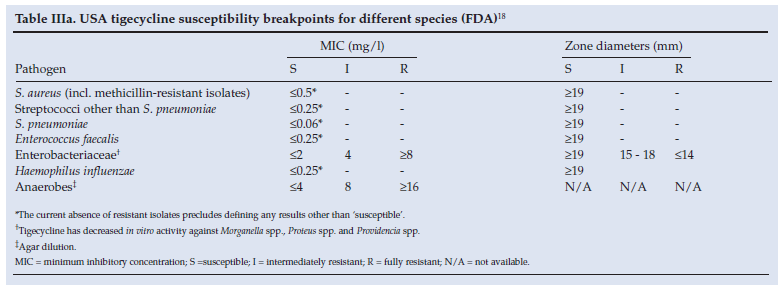
7.1 Breakpoints for tigecycline susceptibility testing
• As depicted in Tables IIIa and IIIb, different breakpoints for Enterobacteriaceae are recommended by different authorities. As a consequence, published susceptibility rates for pneumoniae, E. coli and other Enterobacteriaceae differ depending on which criteria are used.
• Similarly conflicting results and discrepancies between susceptibility methods have raised the question as to which method and which breakpoints should be used when reporting Acinetobacter baumannii susceptibility:15-17
• Most published studies to date have used a provisional breakpoint of <2 mg/l.
• However, the Food and Drug Administration (FDA)18 and the European Committee on Antimicrobial Susceptibility Testing (EUCAST)4 have not made recommendations for A. baumannii, and the British Society for Antimicrobial Chemotherapy (BSAC) has recommended a more conservative breakpoint of susceptibility (<1 mg/l), similar to that of Enterobacteriaceae.19
• Testing for A. baumannii is further complicated by the presence of manganese in Mueller-Hinton media, which can influence susceptibility results (increases in MIC as determined by E-test have been reported).20,21
• Furthermore, with regard to the Enterobacteriaceae, the BSAC recently advised that, owing to a poor correlation between MIC and zone diameters for species other than coli, disc diffusion should not be used and that MIC be determined by E-test. Isolates of E. coli that appear intermediate or resistant on disc testing need confirmation with an MIC.19
7.2 In vitro and in vivo data for tigecycline activity against MDR (including carbapenem-resistant) A. baumannii
• Despite the confusion described above, it is expected that tigecycline will frequently be used in South Africa (alone or in combination) for the treatment of severe A. baumannii infections, particularly in critically ill patients.
• In vitro data derived from 9 of 18 studies reporting on multi-drug resistant (MDR) A. baumannii and 7 of 15 studies reporting specific data on carbapenem-resistant Acinetobacter spp. suggested that at least 90% of strains are susceptible to tigecycline at an MIC breakpoint of <2 mg/l.22
• Tigecycline was also active against 9 of 10 polymyxinresistant strains and 17 of 17 polymyxin intermediateresistant strains.
• These data would, however, represent a serious overestimation of the antimicrobial activity of tigecycline against MDR Acinetobacter spp. if a more conservative breakpoint of <1 mg/l was utilised.
• With regard to clinical efficacy, very few published data are available. Retrospective data compiled from 42 severely ill patients (8 of whom were bacteraemic) treated with tigecycline showed that tigecycline in combination with other antibiotics (mostly polymyxin in 28 patients) was effective in 76% (32/42).22
• Despite clinical efficacy, tigecycline resistance developed during therapy in 3 of the above patients (and was associated with clinical failure in 2). In addition, breakthrough bacteraemia has been reported in 2 patients receiving tigecycline therapy for other infections.22,23 This was not surprising as:
• The MIC90 for A. baumannii in these infections was 2 mg/l, well above the mean peak serum concentration of 0.87 mg/l.
• Acinetobacter spp. are known to have a propensity to acquire resistance, and exposure to sub-therapeutic serum levels may promote its more rapid emergence.
• Clinicians should therefore interpret the in vitro activity of tigecycline cautiously when using it for off-label indications, and treatment should be guided by the MIC.
• The potential for the development of resistance may also be exaggerated by the current registered dose (100 mg loading followed by 50 mg 12-hourly) in critically ill patients, as the higher volume of distribution in these patients may contribute to significant under-dosing (unpublished reports have suggested better efficacy at maintenance doses of 100 - 150 mg 12-hourly).
• Until further data become available, no firm recommendations can be made with regard to the testing or clinical utility of tigecycline, either alone or in combination with polymyxin, the carbapenems (regardless of the mode of delivery), rifampicin (or possibly fosfomycin) for life-threatening MDR Acinetobacter infections. It would, however, seem prudent to use tigecycline at higher doses and in combination with other agents in such infections.
7.3 Tigecycline for bacteraemic patients
• Tigecycline should not be used in primary bacteraemia or in infective endocarditis, as it would appear logical that in these settings the serum concentration should exceed the MIC. Considering that the Cmax is 0.87 mg/l, it would be expected that this agent would not be effective.
• Bacteraemic infections are, however, usually secondary to a primary source and eradication of the source is likely to result in clinical resolution.
• Data from the registration trials in cIAI and cSSTI demonstrated equivalent outcome relative to comparators in patients with concomitant bacteraemia.10,11 It is important to note that no cases of Acinetobacter bacteraemia were reported in these studies and no bacteria had an MIC >1 mg/l.10,11
• Further evidence of efficacy in bacteraemic patients emanated from the trial analysis of CAP due to pneumoniae, and as such tigecycline has been registered by the FDA for CAP caused by S. pneumoniae, including cases with concurrent bacteraemia.18
• Recently, pooled results from 8 phase III clinical trials comparing the safety and efficacy of tigecycline in subjects with secondary bacteraemia were published. Cure rates were similar to comparative standard therapies.24
• The data suggest that tigecycline's extensive tissue distribution allows for eradication of the source of secondary bacteraemia.
• Clinicians should be cautious when using tigecycline for the treatment of patients with suspected or proven bacteraemia, and use should preferably be guided by the MIC.
7.4 In vitro and in vivo data for tigecycline activity against MDR Enterobacteriaceae (ESBL producers or carbapenem-resistant (ertapenem and/or imipenem and/or meropenem) isolates)25
A recent review reported on the in vitro activity of tigecycline against ESBL-producing and carbapenemresistant Enterobacteriaceae (due to either the new pneumoniae carbapenemases or metallo-β-lactamases):
• Tigecycline was active against >99% of MDR E. coli (N= 1 936; of which 1 636 strains were ESBL producers and 14 carbapenem resistant) using either FDA (MIC <2 mg/l) or EUCAST (MIC <1 mg/l) susceptibility criteria.
• Susceptibility rates for MDR K. pneumoniae were 91.2% (N=2 627) and 72.3% (N=1 504) using FDA and EUCAST criteria, respectively. For ESBL-producing isolates susceptibility was 92.3% (N=2 030) and 72.3% (N=1 284) and for carbapenem-resistant strains 94.8% (N=402) and 71.9% (N=231), respectively.
• Susceptibility rates for ESBL-producing Enterobacter spp. were 91.3% (N=69) and 77.6% (N=49), and for carbapenem-resistant strains 80.3% (N=102) and 57.8% (N=102), respectively.
• Clinical efficacy in infections caused by ESBL-producing and carbapenem-resistant Enterobacteriaceae has been reported in 33 patients in 10 studies (cIAI and complicated pelvic infections N=16, bacteraemia N=8, pulmonary infection N=6, urinary tract infection N=3):
• Outcome was favourable in 69.7% of patients (23/33) and classified as uncertain in 3.
• Tigecycline was administered as monotherapy in 23 patients and as combination therapy (mostly together with polymyxin) in 7.
• Of note, two recurrences of empyema occurred in 1 patient with an associated rise in tigecycline MIC (from 0.75 to 2 mg/l).
• Prolonged therapy for microbiological and clinical cure (>21 days) was required in 5 cases.
• It is envisaged that tigecycline will be used in South Africa for directed therapy, particularly for carbapenemresistant strains, where apart from polymyxin and possibly fosfomycin, no alternative Gram-negative antibiotics are available.
7.5 In vitro and in vivo data for tigecycline activity against Stenotrophomonas maltophilia
The majority of clinical isolates of S. maltophilia are inherently resistant to most antimicrobial agents, and as such few therapeutic options remain.
• Recent studies have reported the in vitro activity of tigecycline against this pathogen:
• Tigecycline activity was reported for 131 isolates from patients hospitalised in intensive care units (ICUs) in a multi-centre, multi-national survey and for 108 isolates from ICUs in Canada.26,27
• In both studies tigecycline and trimethoprim/ sulfamethoxazole were the most active agents, with the MIC90 and MIC range reported as 2 and 4 mg/l and 0.12 - 8 and 0.25 - 16 mg/l, respectively. Applying the provisional breakpoint of <2 mg/l used by most studies to date, 90.1% of strains were susceptible.26
• In contrast to the above reports, in which the percentage of isolates cultured from blood was not known, Livermore et al. recently published the susceptibility of S. maltophilia (N=142) isolated from blood cultures in the UK and Ireland; 89% were susceptible at a breakpoint of <1 mg/l, with a reported MIC90 of 1 mg/l and an MIC range of 0.12 - 4 mg/l.28
• With regard to clinical efficacy, few data are available. One case report described successful treatment of a late-onset nosocomial pneumonia caused by an MDR strain of S. maltophilia. 29
7.6 In vitro and in vivo data for tigecycline activity against Clostridium difficile
• Reported MIC90 values for C. difficile are low, ranging from 0.06 to 0.25 mg/l.30,31
• The median faecal concentration of tigecycline is 5.6 mg/l (range 3.0 - 14.1 mg/l) after intravenous administration of a 100 mg loading dose followed by 50 mg twice daily, which is significantly higher than that of metronidazole or its metabolite (median value 0 mg/l, range 0 - 10.2 mg/l).32,33
• It has been demonstrated that tigecycline does not induce proliferation of the organism or enhance cytotoxin production in a human gut model.34
• The intravenous administration of tigecycline is more appealing than oral vancomycin for critically ill patients with C. difficile infection (CDI), as gut mobility is often impaired and in addition it is questionable whether a vancomycin enema can deliver sufficient intra-colonic concentrations, particularly to the transverse and ascending colon.
• Successful treatment of severe refractory CDI has recently been described in case reports in which conventional therapy with metronidazole or vancomycin had failed and colectomy had been considered; all of these patients improved within a week and no relapses were observed.35
8. Appropriate use
Tigecycline has been studied as empiric monotherapy in cIAI, cSSTI and severe CAP and would be an appropriate option as monotherapy for the treatment of patients with cIAI and cSSTI, which are the currently registered indications in South Africa, in the following circumstances:
8.1 Empiric monotherapy
• In the elderly or patients with significant co-morbidity who have received frequent antibiotic therapy or are from long-term care facilities and as such are at risk for resistant bacteria such as ESBL-producing strains or polymicrobial MDR infections (excluding Pseudomonas spp).
• Serious and complicated infections due to MRSA and/or ESBL-producing infections in patients with established renal dysfunction and those at risk of developing renal failure.
• Where there has been treatment failure with other broadspectrum agents despite apparent source control and where pseudomonal infection is unlikely.
• Infections with organisms likely to be susceptible to tigecycline in patients with β-lactam allergy.
• To facilitate heterogeneous antibiotic use and reduce pressure on other agents currently in use as a component of antibiotic stewardship. This might be particularly relevant for the treatment of ESBL-producing Enterobacteriaceae, which has put significant pressure on carbapenems.
8.2 Directed monotherapy
• Polymicrobial infections with MDR organisms excluding Pseudomonas spp., Proteus spp., Providencia spp. and Morganella spp. such as serious and complicated infections due to mixed infections of MRSA or ESBL-producing organisms.
• MRSA infections in the presence of renal dysfunction as an alternative to linezolid. As such, tigecycline might be a treatment option for heterovancomycin intermediate-resistant (h-VISA, MIC >2 mg/l), vancomycin intermediate-resistant (VISA, MIC = 8 mg/l) and vancomycin-resistant (VRSA, MIC >16 mg/l) aureus infections as well as VRE infections.
8.3 Directed combination therapy
• It is uncertain whether tigecycline will be effective with more resistant Acinetobacter spp. infections (MICs >2 mg/l). However, if no other antibiotic is available according to susceptibility testing, it may be utilised as salvage therapy in combination with other agents. Combinations with polymyxin and/or fosfomycin and/or rifampicin have mostly been reported, but as has been stated, insufficient data exist to make firm recommendations.
• If used in these circumstances, pharmacokinetic/pharmacodynamic data suggest that it should be used at higher than the registered dose.
9. Inappropriate use
9.1. Tigecycline is not an appropriate empiric option as monotherapy for the treatment of patients with cIAI at risk of infection with P. aeruginosa, and in particular those with recurrent infection and/or failure of source control.36-39
9.2 Tigecycline is not an appropriate empiric monotherapy option for the treatment of patients with cSSTI where P. aeruginosa is a predominant organism, such as chronic diabetic foot infection.40
10. Conclusion
This statement has been developed to promote the appropriate use of tigecycline in SSTIs and IAIs, which are the current registered indications for this antibiotic in South Africa. In the USA tigecycline is also registered for CAP, and when this indication is registered in South Africa, the statement will be updated. Studies are also currently ongoing for other indications and in children.
11. References
1. Pankey GA. Tigecycline. J Antimicrob Chemother 2005; 56: 470-480 doi:10.1093/jac/dki248. [ Links ]
2. Peterson LR. A review of tigecycline: the first glycylcyclines. Int J Antimicrob Agents 2008; 32: S215-S222 doi:10.1016/S0924-8579(09)70005-6. [ Links ]
3. Rello J. Pharmacokinetics, pharmacodynamics, safety and tolerability of tigecycline. J Chemother 2005; 17: 12-22. [ Links ]
4. EUCAST technical note on tigecycline. Clin Microb Infect 2006; 12: 1147-1149 doi: 10.1111/ j.1469-0691.2006.01578.x. [ Links ]
5. Nathwani D. Tigecycline: clinical evidence and formulary positioning. Int J Antimicrob Agents 2005; 25: 185-192. [ Links ]
6. van Ogtrop ML, Andes D, Stamstad TJ, et al. In vivo pharmacodynamic activities of two glycylcyclines (GAR-936) AND way 152,288) against various Gram-positive and Gramnegative bacteria. Antimicrob Agents Chemother 2000; 44: 943-949. [ Links ]
7. Hoban DJ, Bouchillon SK, Johnson BM, Johnson JL, Dowzicky MJ. Tigecycline Evaluation and Surveillance Trial (TEST program) Group. In vitro activity of tigecycline against 6792 Gram-negative and Gram-positive clinical isolates from the global Tigecycline Evaluation and Surveillance Trial (TEST program 2004). Diagn Microbiol Infect Dis 2005; 52: 215-217 doi:10.1016/j.diagmicrobio.2005.06.001. [ Links ]
8. Reinert RR, Low DE, Rossi F, Zhang X, Wattal C, Dowzicky MJ. Antimicrobial susceptibility among organisms from Asia/Pacific Rim, Europe and Latin and North America collected as part of TEST and the in vitro activity of tigecycline. J Antimicrob Chemother 2007; 60: 1018-1029 doi:10.1093/jac/dkm310. [ Links ]
9. Entenza JM, Moreillon P. Tigecycline in combination with other antimicrobials: a review of in vitro, animal and case report studies. Int J Antimicrob Agents 2009; 34: 8, e1-e9 doi:10.1016/j. ijantimicag.2008.11.006. [ Links ]
10. Babinchak T, Ellis-Grosse E, Dartois N, Rose GM, Loh E. Tigecycline 301 study group; Tigecycline 306 study group. The efficacy and safety of tigecycline for the treatment of complicated intra-abdominal infections: analysis of pooled clinical trial data. Clin Infect Dis 2005; 41: S354-367 doi: 10.1086/431676. [ Links ]
11. Ellis-Grosse EJ, Babinchak T, Dartois N, Rose GM, Loh E. Tigecycline 300 cSSSI study group; Tigecycline 305 cSSSI study group. The efficacy and safety of tigecycline in the treatment of skin and skin-structure infections: results of 2 double-blind phase 3 comparison studies with vancomycin-aztreonam. Clin Infect Dis 2005; 41: S341-353 doi: 10.1086/431675. [ Links ]
12. Tanaseanu C, Bergallo C, Teglia O, et al. Integrated results of 2 phase 3 studies comparing tigecycline and levofloxacin in community-acquired pneumonia. Diagn Microbiol Infect Dis 2008; 61: 329-338 doi:10.1016/j.diagmicrobio.2008.04.009. [ Links ]
13. Vasilev K, Reshedko G, Orasan R, et al. A phase 3, open-label, non-comparative study of tigecycline in the treatment of patients with selected serious infections due to resistant Gramnegative organisms including Enterobacter species, Acinetobacter baumannii and Klebsiella pneumoniae. J Antimicrob Chemother 2008; 62: i29-i40 doi:10.1093/jac/dkn249. [ Links ]
14. Florescu I, Beuran M, Dimov R, et al. Efficacy and safety of tigecycline compared with vancomycin or linezolid for treatment of serious infections with methicillin-resistant Staphylococcus aureus or vancomycin-resistant enterococci: a Phase 3, multicentre, doubleblind, randomized study. J Antimicrob Chemother 2008; 62: i17-i28 doi:10.1093/jac/dkn250. [ Links ]
15. Navon-Venezia S, Leavitt A, Carmeli Y. High tigecycline resistance in multidrug-resistant Acinetobacter baumannii. J Antimicrob Chemother 2007; 59: 772-774 doi:10.1093/jac/dkm018. [ Links ]
16. Thamlikitkul V, Tiengram S, Tribuddharat C. Comment on: High tigecycline resistance in multidrug-resistant Acinetobacter baumannii. J Antimicrob Chemother 2007; 59(4): 772-774 doi.1093/jac/dkm142. [ Links ]
17. Jones RN, Ferraro MJ, Reller LB, Schrenberger PC, Swenson J, Sader HS. Multicentre studies of tigecycline disk diffusion susceptibility results for Acinetobacter spp. J Clin Microbiol 2007; 35: 227-230 doi: 1128/JCM.01588-06. [ Links ]
18. United States Food and Drug Administration (FDA), Highlights of prescribing information Tygacil. http://www.fda.gov/Safety/MedWatch/SafetyInformation/SafetyRelatedDrugLabelingChanges/ucm132714.htm (accessed 6 April 2010). [ Links ]
19. BSAC Methods for Antimicrobial Susceptibility Testing Version 8. January 2009. hhtp://www.bsac.org.uk/susceptibility_testing/bsac_standardized_disc_susceptibility_method.cfm (accessed 31 August 2009). [ Links ]
20. Casal M, Rodriguez F, Johnson B, et al. Influence of testing methodology on the tigecycline activity profile against presumably tigecycline-non-susceptible Acinetobacter spp. J Antimicrob Chemother 2009; 64: 69-72 doi:10.1093/jac/dkp169. [ Links ]
21. Fernandez-Mazarrasa C, Mazarrasa O, Calvo J, del Arco A, Martinez-Martinez L. High concentrations of manganese in Mueller-Hinton agar increase MICs of tigecycline determined by E-test. J Clin Microbiol 2009; 47: 827-829 doi:10.1128/JCM.02464-08. [ Links ]
22. Karageorgopoulos DE, Kelesedis T, Kelesedis I, Falagas ME. Tigecycline for the treatment of multidrug-resistant (including carbapenem resistant) Acinetobacter infections: a review of the scientific evidence. J Antimicrob Chemother 2008; 62: 45-55 doi:10.1093/jac/dkn165. [ Links ]
23. Peleg AY, Potoski BA, Rea R, et al. Acinetobacter baumannii bloodstream infection while receiving Tigecycline: a cautionary report. J Antimicrob Chemother 2007; 59:128-131 doi:10.1093/jac/dkl441. [ Links ]
24. Gardiner D, Dukart G, Cooper C, Babinchak T. Safety and efficacy of intravenous tigecycline in subjects with secondary bacteraemia: pooled results from 8 phase III clinical trials. Clin Infect Dis 2010; 50: 229-38 doi: 10.1086/648720. [ Links ]
25. Kelesedis T, Karageorgopoulos DE, Kelesedis I, Falagas ME. Tigecycline for the treatment of multidrug-resistant Enterobacteriaceae: a systematic review of the evidence from microbiological and clinical studies. J Antimicrob Chemother 2008; 62: 895-904 doi:10.1093/ jac/dkn311. [ Links ]
26. Sader HS, Jones RN, Dowzicky MJ, Fritsche TR. Antimicrobial activity of tigecycline tested against nosocomial bacterial pathogens from patients hospitalized in the intensive care unit. Diagn Microbiol Infect Dis 2005; 52: 203-208 doi:1016/j.diagmicrobio.2005.05.002. [ Links ]
27. Zhanel GG, DeCorby M, Nichol KA, et al. Antimicrobial susceptibility of 3931 organisms isolated from intensive care units in Canada: Canadian National Intensive Care Unit Study, 2005/2006. Diagn Microbiol Infect Dis 2008; 62: 67-80 doi:10.1016/j.diagmicrobio.2008.04.012. [ Links ]
28. Livermore DM, Hope R, Brick G, et al. Non-susceptibility trends among Pseudomonas aeruginosa and other non-fermentative Gram-negative bacteria from bacteraemias in the UK and Ireland, 2001-06. J Antimicrob Chemother 2008; 62: ii55-ii63 doi:10.1093/jac/dkn352. [ Links ]
29. Blanquer D, De Otoro J, Padilla E, et al. Tigecycline for treatment of nosocomial-acquired pneumonia possibly caused by multi-drug resistant strains of Stenotrophomonas maltophilia. J Chemother 2008; 20: 761-763. [ Links ]
30. Baines SD, Saxton K, Freeman J, Wilcox MH. Tigecycline does not induce proliferation or cytotoxin production by epidemic Clostridium difficile strains in a human gut model. J Antimicrob Chemother 2006; 58: 1062-1065 doi:10.1093/jac/dkl364. [ Links ]
31. Hecht DW, Galang MA, Sambol SP, Osmolski JR, Johnson S, Gerding DN. In vitro activities of 15 antimicrobial agents against 110 toxigenic Clostridium difficile clinical isolates collected from 1983 to 2004. Antimicrob Agents Chemother 2007; 51: 2716-2719 doi:10.1128/AAC.01623-06. [ Links ]
32. Bolton RP, Culshaw MA. Faecal metronidazole concentrations during oral and intravenous therapy for antibiotic associated colitis due to Clostridium difficile. Gut 1986; 27: 1169-1172 doi:10.1136/gut.27.10.1169. [ Links ]
33. Nord CE, Sillerstrom E, Wahlund E. Effect of tigecycline on normal oropharyngeal and intestinal microflora. Antimicrob Agents Chemother 2006; 50: 3375-3380 doi:10.1128/ AAC.00373-06. [ Links ]
34. Baines SD, Saxton K, Freeman J, Wilcox MH. Tigecycline does not induce proliferation or cytotoxin production by epidemic Clostridium difficile strains in a human gut model. J Antimicrob Chemother 2006; 58: 1062-1065 doi:10.1093/jac/dkl364. [ Links ]
35. Herpers BL, Vlaminckx B, Burkhardt O, et al. Intravenous tigecycline as adjunctive or alternative therapy for severe refractory Clostridium difficile infection. Clin Infect Dis 2009; 48: 1732-1735 doi: 10.1086/599224. [ Links ]
36. Farthmann EH, Schöffel U. Epidemiology and pathophysiology of intraabdominal infections (IAI). Infection 1998; 26: 329-334 doi: 10.1007/BF02962266. [ Links ]
37. LaRoche M, Harding G. Primary and secondary peritonitis: an update. Eur J Clin Microbiol Infect Dis 1998; 17: 542-550 doi: 10.1007/BF01708616. [ Links ]
38. Malangoni MA. Evaluation and management of tertiary peritonitis. Am Surg 2000; 66: 157-161 [ Links ]
39. Barie PS. Management of complicated intra-abdominal infections. J Chemother 1999; 11: 464-477. [ Links ]
40. Lipsky BA, Berendt AR, Deery HG, et al. Diagnosis and treatment of diabetic foot infections. Clin Infect Dis 2004; 39: 885-910 doi: 10.1086/424846. [ Links ]
12. Acknowledgements
The Working Group meeting was sponsored through an unrestricted educational grant from Wyeth. Representatives from the company attending the meeting had observer status.
13. Endorsement
Endorsed by the Association of Surgeons of South Africa, the Trauma Society of South Africa, the Federation of Infectious Diseases Societies of Southern Africa, the Critical Care Society of Southern Africa and the South African Thoracic Society.
14. Disclaimer
This statement is published for educational purposes only. The recommendations are based on currently available scientific evidence together with the consensus opinion of the authors.
15. Working group
A J Brink (Ampath National Laboratory Services, Milpark Hospital, Johannesburg), D Bizos (Acting Head, Surgical Gastro-enterology, Department of Surgery, Charlotte Maxeke Johannesburg Academic Hospital and University of the Witwatersrand, Johannesburg), K D Boffard (Chief Specialist and Head, Department of Surgery, Charlotte Maxeke Johannesburg Academic Hospital and University of the Witwatersrand, Johannesburg), C Feldman (Division of Pulmonology, Department of Medicine, Charlotte Maxeke Johannesburg Academic Hospital and University of the Witwatersrand, Johannesburg), D C Grolman (Specialist Surgeon and Intensivist, Sandton Medi-Clinic, Life Fourways Hospital and University of the Witwatersrand, Johannesburg), J P Pretorius (Surgical and Trauma ICU, Department of Surgery, Steve Biko Academic Hospital, University of Pretoria), G A Richards (Director Critical Care and Principal Physician, Division of Pulmonology, Department of Medicine, Charlotte Maxeke Johannesburg Academic Hospital and University of the Witwatersrand, Johannesburg), M Senekal (Pathcare, Panorama Mediclinic, Cape Town), E Steyn (Trauma and Transplant Surgeon, Christiaan Barnard Memorial Hospital, Cape Town), N Welkovic (Specialist Surgeon and Intensivist, Unitas Hospital and Division of Critical Care, Steve Biko Academic Hospital, University of Pretoria).
Please forward all comments to: Dr A J Brink, PO Box 1873, Houghton, 2041, tel. (011) 726-6260, e-mail brinka@ampath.co.za


{kind=link}
{kind=link}
{kind=link}