










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSAMJ: South African Medical Journal
On-line version ISSN 2078-5135
Print version ISSN 0256-9574
SAMJ, S. Afr. med. j. vol.99 n.6 Pretoria Jun. 2009
ORIGINAL ARTICLES
Cerebral autosomal dominant arteriopathy with subcortical infarcts and leucoencephalopathy (CADASIL)
Chris RetiefI; Clara-Maria SchutteII, *; Malcolm Kevin BakerIII
IMB ChB, MMed (Neurol), Department of Neurology, University of Pretoria
IIMB ChB, MMed (Neurol), MD, Department of Neurology, University of Pretoria
IIIMB ChB, MMed (Neurol), FCP MD (USA), DABPN, Department of Neurology, University of Pretoria
ABSTRACT
BACKGROUND: Cerebral autosomal dominant arteriopathy with subcortical infarcts and leuco-encephalopathy (CADASIL) is a hereditary autosomal dominant non-atherosclerotic non-amyloid cerebral arteriopathy. The disease was identified in 1993. We are not aware of reports in the literature of its occurrence in South Africa, and we present the clinical and laboratory features of 5 patients with CADASIL.
METHODS: Patients with the characteristic radiological white matter disease and typical features (family history, ischaemic events, migraine or dementia) were evaluated for possible CADASIL by means of clinical examination, routine investigations for strokes, magnetic resonance imaging, skin biopsy electron microscopy, evoked potentials and electro-encephalography.
RESULTS: The clinical and laboratory features of our study largely correlate with reported studies. However, all of the skin biopsies were positive, and the onset of migraine in our patients was considerably earlier. A new finding, to our knowledge, was the normality of visual, somatosensory and auditory evoked potentials.
CONCLUSION: Our study confirms the existence of CADASIL in South Africa, and also suggests that skin electron microscopy is useful, despite recent reports of its low sensitivity, and that evoked potentials in CADASIL are likely to be normal.
Cerebral autosomal dominant arteriopathy with subcortical infarcts and leuco-encephalopathy (CADASIL) is a progressive hereditary non-atherosclerotic non-amyloid arteriopathy first described in a French pedigree, affecting young and older adult patients irrespective of traditional risk factors for stroke.1 The French group, who also localised the abnormal gene to chromosome 19 in their affected patients, coined the term CADASIL.2 The specific abnormality is a mutation of the Notch3 gene, causing damage to vascular smooth-muscle cells. Clinically, patients may present with strokes, migraine, progressive dementia and mood disorders. CADASIL has not been described in South Africa, and we report on 5 South African patients with the condition.
The patients were referred for specialist opinion and were evaluated at the Neurology Department of Pretoria Academic Hospital. No informed consent was obtained since they were seen as part of routine neurology practice and are reported retrospectively. Patients were clinically evaluated, and standard laboratory investigations, echocardiography, carotid Dopplers, electro-encephalography and evoked potential studies, magnetic resonance imaging (MRI) of the brain and full-thickness skin biopsies were performed.
Patients and methods
Case 1
A 60-year-old white woman presented with progressive left hemiparesis, similar to an episode she had experienced 2 years previously but that had resolved spontaneously. Her medical history included mild hypertension controlled by perindopril, and hypothyroidism responding to 0.1 mg of thyroxine. Several individuals in her family had suffered strokes over at least 3 generations. General examination and cognition were normal. She had a mild left hemiparesis with bilaterally brisk reflexes (more on the left), with unsustained clonus and a positive Hoffmann reflex on the left; the tone was increased bilaterally, but sensory and cerebellar examination were normal.
Special investigations, including lipogram, glucose, full blood count, anti-nuclear factor, rheumatoid factor, syphilis serology, HIV serology, protein S, protein C, antithrombin III, activated protein C resistance index, lupus anticoagulant, cardiolipin antibodies, homocysteine, ECG, chest X-rays, transthoracic echocardiography and carotid Doppler, were normal.
MRI of the brain showed widespread white matter abnormalities, especially in the regions of the anterior temporal lobes and the external capsules. Lacunar infarcts were seen in the right lentiform nucleus and thalamus and in the superficial white matter. On diffusion imaging, a hyperintense signal area in the right corona radiata was noted, probably the cause of the recent left hemiparesis (Fig. 1).
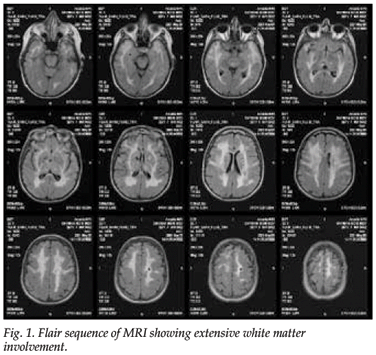
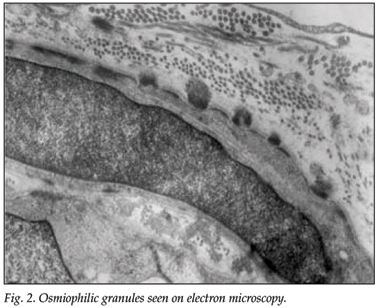
Electron microscopy (EM) of a full-thickness skin biopsy revealed granular osmiophilic material (GOM) embedded within the basement membranes of several arterioles, indenting the cell membranes of the vascular smooth-muscle cells, and fragmentation of the vascular smooth-muscle cells and thickening of the basal membranes (Fig. 2).
The visual and auditory evoked potentials and an EEG were normal. Nerve conduction studies of the lower limbs showed no evidence of a polyneuropathy.
Case 2
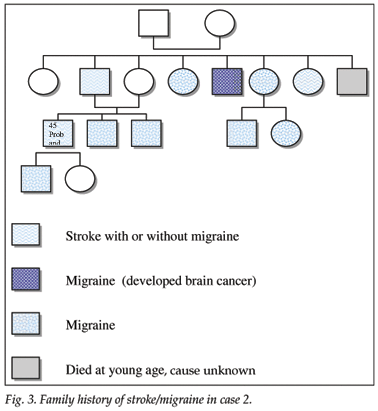
A 45-year-old white man complained of episodic paraesthesiae of his right hand and speech impairment, followed by severe headache, occurring since the age of 27 years. A neurologist saw him in 2001. The patient had a right-sided hemiparesis and mild asthma, but no stroke risk factors. Two close family members had had strokes, and at least 8 family members had a positive history of migraine (Fig. 3).
At the time of the stroke, general examination and blood pressure were normal, and a right hemiplegia and right hemisensory deficit with slurred speech were found. The patient was seen at Pretoria Academic Hospital 2 years after this incident, with normal cognition, residual weakness on the right side, and bilateral upper motor neuron signs, more marked on the right side. He had a raised total cholesterol level of 7.5 mmol/l.
An MRI scan of the brain showed widespread white matter lesions with moderate involvement of the anterior temporal lobes and extensive lesions in the external capsule. There was hyperintensity of the posterior left lentiform nucleus/posterior limb of the internal capsule and a small lacuna at the cortical-subcortical junction in the left hemisphere.
Electron microscopy showed an abundance of osmophilic granules; the visual, auditory and somatosensory evoked potentials were normal, as was the EEG.
Case 3
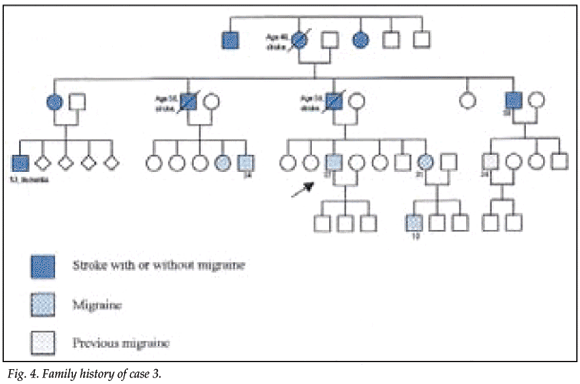
A 37-year-old white man presented with migraine, which had started during childhood and worsened after a head injury. The head injury had resulted in loss of consciousness for 5 hours and anterograde amnesia for 2 days. The migraines commenced with a visual aura followed by severe unilateral headache; he experienced an unusually severe attack in 2002, with clouding of consciousness and amnesia lasting 3 days, followed by intermittent left-sided paraesthesiae and visual disturbances in the left field of vision. Seven members of his family had suffered strokes, and at least 5 others complained of migraine (Fig. 4). His blood pressure and general and neurological examinations were normal, as was cognition.
Laboratory tests showed a normal cholesterol level of 4.7 mmol/l, but high-density lipoprotein was slightly decreased at 0.7 mmol/l and low-density lipoprotein was increased (35 mmol/l). No Doppler studies were done.
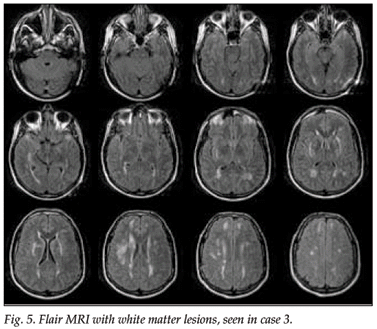
An MRI scan of the brain showed widespread white matter involvement with prominent abnormalities in the external capsules and to a lesser extent in the anterior temporal lobes.
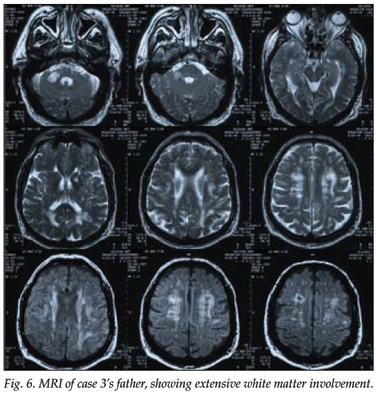
An MRI scan of the patient's father, who had had a similar history and had died at the age of 58, also showed marked white matter lesions (Figs 5 and 6).
Electron microscopy of a full-thickness skin biopsy showed scattered osmiophilic granules in the basement membranes of several vascular smooth-muscle cells.
The visual, auditory and somatosensory evoked potentials, and the EEG were within normal limits.
Case 4
A 46-year-old white man complained of episodic visual auras consisting of fortification spectra and hemianopia that had started in early adulthood. Headache was initially not a prominent feature. During the last decade, he had experienced hypo- and dysaesthesiae of half of his face and ipsilateral hand together with a visual aura, followed by severe headache. Symptoms cleared over 2 days and recurred approximately monthly. In 2002, he experienced a sudden monoplegia of the right arm which was not accompanied by the other symptoms, which also cleared up gradually.
His medical history included hypertension, which was controlled on perindopril. He had never smoked. One family member had had a stroke, but the patient could not supply much information on relatives. His blood pressure and cognition were normal; mild weakness was present in the right hand, and reflexes were brisk bilaterally. Pronator teres tone was increased on the right, and he had a mild sensory loss over the right half of the face and right arm. He had an elevated total cholesterol level (6.6 mmol/l) and cardiolipin IgM antibodies were borderline raised (22.4 units). Echocardiography and Doppler of the carotids were normal.
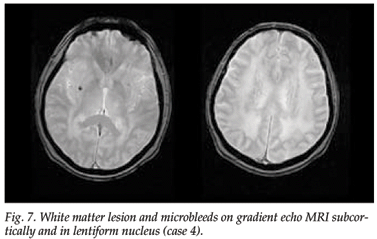
MRI scanning showed extensive diffuse white matter lesions, with involvement of the anterior temporal poles and marked involvement of the external capsule, and a large lacuna in the pons surrounded by hyperintensities. A gradient-echo MRI scan showed two microbleeds, one subcortically and one in the lentiform nucleus (Fig. 7).
Electron microscopy of the skin biopsy showed proliferation of vascular smooth-muscle cells, crenation of the internal elastic lamina and fragmentation of elastic fibres. The basement membrane was irregular and osmiophilic granules were observed.
Neurophysiological examination showed normal visual, auditory and somatosensory evoked potentials, while the EEG revealed a single focal burst of sharp transients left mid-temporally; the alpha rhythm showed lower voltage on the left side.
Case 5
A 67-year-old white woman presented with memory problems and behavioural disturbances. The short-term memory loss had commenced 3 years previously; the behavioural disturbances were more recent, involving paranoid delusions, inappropriate behaviour and an obsession with sweets and diet pills. Episodes of confusion and a gait disturbance involving stiffness of the right leg had been noticed. Previously, the patient had suffered from migraine and had been treated for depression. She did not smoke. At least 2 other family members had suffered from strokes; one had been diagnosed with dementia. The patient's daughter suffered from migraine.
Blood pressure and general examination were normal; cognition was impaired, with a Mayo Mini-mental Score of 28/38. Primitive reflexes (snout reflex, palmomental reflex) were present. On the right side, there was mild weakness of the hand and toe extension, brisker reflexes and mild spasticity. Both plantar responses were extensor and Hoffmann reflexes were present on both sides. Both hands were clumsy and the gait was broad-based with a tendency to invert the right foot.
Renal, liver and thyroid functions, full blood count, glucose and vitamin B12 were normal. The antinuclear factor was 1:40, and the rheumatoid factor was negative. Cerebrospinal fluid examination showed only a mild increase in protein (578 mg/l). An MRI scan showed widespread white matter lesions, particularly prominent in the anterior temporal regions and external capsule. Multiple small linear lacunes were seen subcortically. A gradient-echo MRI showed several microbleeds.
Skin biopsy electron microscopy showed typical osmiophilic granules. Neurophysiological investigations showed normal auditory and somatosensory evoked potentials with a normal visual evoked potential on the right and borderline abnormality on the left (prolonged latencies). The EEG was abnormal, with episodes of 2 - 3 Hz delta in the frontal regions.
Discussion
CADASIL is an autosomal dominant widespread arteriopathy, which may present with migraine, ischaemic central nervous system lesions (transient ischaemic attacks/strokes), subcortical dementia, and mood disorders.
Our patients had a positive family history of strokes in first-degree relatives, except for one who was unable to provide family details. This is in keeping with an autosomal dominant form of inheritance. Migraine with aura was present in 3 patients in whom it was also the first symptom of the disorder; complicated migraine occurred in 1 patient (case 2); case 3 reported a severe migraine, which was followed by clouding of consciousness and amnesia – an episode difficult to distinguish from an ischaemic event. Migraine started before the age of 30 years in all patients, in keeping with other reports.3-5 The mechanisms causing migraine in CADASIL are unclear – functional alterations of small cerebral arteries may be involved.
Ischaemic events occurred in all our patients, with the first stroke seen at a mean age of 50 years (37 - 66 years). They recovered well after the ischaemic incident; all but one were fully functional after a few months. Traditional vascular risk factors were present in 3 patients, with 2 showing hypercholesterolaemia and 1 hypertension.
The MRI scans showed involvement of the anterior temporal lobes and external capsule in all patients, as typically stated in other reports.6,7 Microbleeds, seen in at least 2 of our patients, were recently also described in patients with CADASIL, occurring in up to 50% of cases.8 Cognitive changes were seen in about 60% of symptomatic individuals, including episodic memory deficits, attention problems, and executive and visuospatial functions and psychomotor slowing.4,5 One would expect about two-thirds of cases, by the age of 65 years, to have evidence of dementia; only 1 of our patients (67 years old) experienced problems with cognition, probably as the others were relatively young.
Mood disorders occur in about 30% of patients with CADASIL.4,5 One of our patients had a previous diagnosis of dysthymia, progressing to behavioural disturbances and dementia within a short timespan.
Few neurophysiological studies on patients with CADASIL have been reported. Sensory neuropathy was found in 7 out of 11 patients who had signs of peripheral nerve involvement.9 Chabriat et al. reported normal nerve conduction studies on 5 of their patients.4 Our patient who had nerve conduction studies was normal. No reports on somatosensory and auditory evoked potentials studies in patients with CADASIL were found. Parisi et al. reported visual function impairment studies in some patients.10 All our patients had normal results for visual, auditory and somatosensory evoked potentials (one showed slight prolongation of latency of the visual evoked potential (VEP)), contrasting with other conditions where CNS white matter disease is prominent. Electroencephalographic studies have also not been reported widely, although up to 10% of CADASIL patients may develop epileptic seizures. Two of our patients had abnormal EEGs: in one, sharp waves in the temporal region raised the possibility of an ictal phenomenon although the patient was asymptomatic, and the patient with dementia showed frontal delta activity on her EEG.
Skin biopsies on electron microscopy in patients with CADASIL show a specificity of close to 100% and a sensitivity of 45 - 96%.11-13 All our patients had an abundance of osmiophilic granules on electron microscopy of their skin biopsies, which confirmed our diagnosis of CADASIL.
Genetic studies, which are not yet available in South Africa, could not be carried out owing to the logistics of tissue transport and costs.
Conclusion
CADASIL occurs in white South Africans, which is not unexpected as many ancestors originate from the countries where CADASIL was first described (e.g. France). A family history must be taken in younger-onset stroke, migraine and dementia patients to correctly identify familial stroke syndromes such as CADASIL and CARASIL (cerebral autosomal recessive arteriopathy with subcortical infarcts and leucoencephalopathy, or Maede syndrome). All our clinically suspected cases showed positive electron microscopy results, and MRI was useful in making the preliminary diagnosis. The observation that the evoked potentials studies were normal is notable and may be of value in differentiating CADASIL from other CNS demyelinating diseases.
Professor L Dreyer of the Anatomical Pathology Department, University of Pretoria, and Professor P R Bartel of the Neurophysiology Unit are thanked for their assistance.
References
1. Sourander P, Walinder J. Hereditary multi-infarct dementia. Morphological and clinical studies of a new disease. Acta Neuropathol (Beli) 1977; 39(3): 247-254. [ Links ]
2. Tournier-Lasserve E, Joutel A, Melki J, et al. Cerebral autosomal dominant arteriopathy with subcortical infarcts and leucoencephalopathy maps to chromosome Igq 19. Nat Genet 1993; 3(3): 256-259. [ Links ]
3. Desmond DW, Moroney JT, Lynch T, Chan S, Chin SS, Mohr JP. The natural history of CADASIL: a pooled analysis of previously published cases. Stroke 1999; 30(6): 1230-1233. [ Links ]
4. Chabriat H, Vahedi K, Iba-Zizen MT, et al. Clinical spectrum of CADASIL: a study of 7 families. Lancet 1995; 346(8980): 934-939. [ Links ]
5. Dichgans M, Mayer M, Uttner I, et al. The phenotypic spectrum of CADASIL: clinical findings in 102 cases. Ann Neurol 1998: 44(5): 731-739. [ Links ]
6. O'Sullivan M, Jarosz JM, Martin RJ, Deasy N, Powell JF, Markus HS. MRI hyperintensities of the temporal lobe and external capsule in patients with CADASIL. Neurology 2000; 56(5): 628-634. [ Links ]
7. Van den Boom R, Lesnik Oberstein SA, Ferrari MD, Haan J, Van Buchem MA. CADASIL: MR imaging findings at different ages – 3rd - 6th decades. Radiology 2003; 229(3): 683-690. [ Links ]
8. Dichgans M, Hoffmannspotter M, Herzog J, Peters N, Bergmann M, Yousry TA. Cerebral microbleeds in CADASIL: a gradient echo MRI and autopsy study. Stroke 2002; 33(1): 67-71. [ Links ]
9. Sicurelli F, Dotti MT, Destetano N, et al. Peripheral neuropathy in CADASIL. J Neurol 2005; 252(10): 1206-1209. [ Links ]
10. Parisi V, Pierelli F, Fattapposta F, et al. Early visual function impairment in CADASIL. Neurology 2003; 60(12): 2008-2010. [ Links ]
11. Markus HS, Martin RJ, Simpson MA, et al. Diagnostic strategies in CADASIL. Neurology 2002; 59(8): 1134-1138. [ Links ]
12. Joutel A, Favrole P, Labauge P, et al. Skin biopsy immunostaining with a Notch3 monoclonal antibody for CADASIL diagnosis. Lancet 2001; 358(9298): 2049-2051. [ Links ]
13. Lesnik Oberstein SA, Van Duinen SG, Van den Boom R, et al. Evaluation of diagnostic NOTCH3 immunostaining in CADASIL. Acta Neuropathol (Berl) 2003; 106(2): 107-111. [ Links ]
Accepted 5 January 2009.
* Corresponding author: Clara-Maria Schutte (cschutte@medic.up.ac.za)

