










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSAMJ: South African Medical Journal
On-line version ISSN 2078-5135
Print version ISSN 0256-9574
SAMJ, S. Afr. med. j. vol.99 n.3 Pretoria Mar. 2009
GUIDELINE
Guideline for using growth hormone in paediatric patients in South Africa: treatment of growth hormone deficiency and other growth disorders
David Segal
ABSTRACT
The Paediatric and Adolescent Endocrine and Diabetes Society of South Africa (PAEDS-SA) recommends, in line with other international groups, that growth hormone (GH) therapy be considered for children and adolescents with significantly short stature and poor growth velocity in the following instances: GH deficiency; Turner syndrome; Prader-Willi syndrome; small-for-gestational-age children with failure of catch-up growth; idiopathic short stature; and chronic renal insufficiency. We have produced treatment guidelines for the use of GH, designed to allow flexibility to determine coverage on a case-by-case basis. We further recommend that when used for growth promotion, GH therapy should be initiated and monitored by, or in consultation with, a paediatric endocrinologist.
1. Introduction
The diagnosis and management of growth disorders in children, particularly conditions that respond to growth hormone (GH), raises challenging clinical and economic issues. This document is intended to provide guidelines for the investigation of children with short stature, together with evidence-based recommendations for the use of GH therapy, where appropriate. Consideration has been given to producing guidelines authored by paediatric endocrinologists that can assist payers in both the private and public sectors to make coverage decisions consistent with endocrine treatment around the world, as practised by paediatric endocrinologists involved with the Paediatric and Adolescent Endocrine and Diabetes Society of South Africa (PAEDS-SA), a professional society affiliated to the Society for Endocrinology, Metabolism and Diabetes of South Africa (SEMDSA).
Growth in humans is a very complex biological process. Historically, GH treatment was restricted to children with proven GH deficiency. However, since the introduction of synthetic GH in 1985, other conditions associated with poor growth have been treated successfully with GH. Currently accepted evidence-based indications for GH are:
• GH deficiency
• Turner syndrome
• Prader-Willi syndrome
• Small-for-gestational-age (SGA) children who fail to show catch-up growth
• Idiopathic short stature
• Chronic renal failure.
This guideline briefly summarises each of these indications and gives evidence-based recommendations for treatment.
2. Evaluation and diagnosis of the child with short stature
2.1 Stature
Short stature is a statistical and a relative term. A percentage of normal individuals will, by definition, be below the 3rd percentile for height in any given population, and heights for different population groups around the world differ considerably. In most cases population-specific growth charts are not available and 'standardised' growth charts, such as those provided by the Centers for Disease Control and Prevention (Atlanta, GA, USA), available at http://www.cdc.gov/GROWTHCHARTS/, are used as a benchmark across multiple population groups. However, patient demographics and ancestry need to be taken into account when using standardised charts.
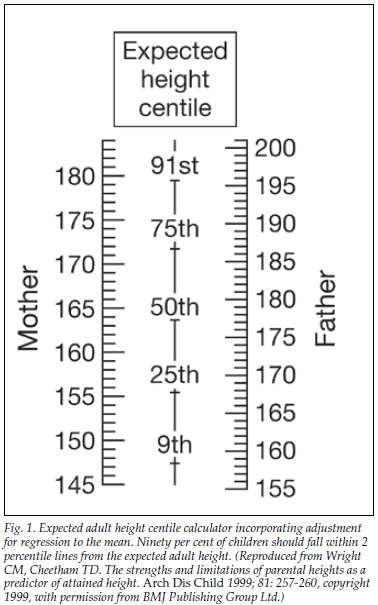
An estimate of the final height of an individual, corrected for genetics, can be obtained by calculating mid-parental height (MPH). This is determined by averaging the height of both parents and adding 7 cm for males or subtracting 7 cm for females. The final height of an individual would be expected to fall within 1.5 standard deviation scores (SDS) or 2 percentile lines above or below the MPH percentile.1 Fig. 1 shows an expected height centile calculator devised by Wright and Cheetham.1 Even if children are within 'normal' growth centiles, growth problems may be present if individuals are below their genetic potential.
It is impossible to begin to manage what we cannot measure, and for this reason it is imperative that children are measured when they come into contact with health care providers such as clinic sisters, general practitioners or paediatricians. Data from all children should be plotted on age- and sex-appropriate growth charts.
2.2 Measurement technique
Height should be measured with an appropriate stadiometer device (Fig. 2). The child should stand with the heels, buttocks and occiput touching the wall, and the eyes and ears in the Frankfurt plane (i.e. the tragus of the ear on the same horizontal plane as the outer canthus of the eye). An average of three measurements (done at the same time) will improve accuracy. Ideally, follow-up measurements should be performed by the same person on the same equipment, and plotted on an appropriate height chart.

Length measurements (Fig. 3) are difficult to perform without appropriate staff and equipment, but correctly obtained data should be plotted on a 0 - 36-month length chart. Children are longer than they are tall, and attention needs to be paid when transitioning or comparing lengths and heights, especially for individuals who are between 2 and 3 years of age.
A valuable additional tool is the growth velocity chart, which enables the average rate of growth to be monitored for a child's age and sex. Fig. 4 shows a height velocity chart for boys, published by Tanner and Davies.2 Using such instruments, a growth rate (the number of centimetres grown per year) that falls below the 25th percentile indicates that catch-up growth is not occurring; growth velocities below the 10th percentile indicate a child who will fall further away from the current height percentile.

All children growing significantly below either the 3rd percentile or their genetic potential (more than 2 percentile spaces, or 1.5 SDS, below their MPH centile), or children whose growth measurements have fallen from their previous growth percentile, should be evaluated for causes of poor growth. Childhood is the only time available for growth, and delaying investigation and management of growth disorders is liable to result in poorer outcomes with regard to final height. Action should not be deferred until puberty in the hope that catch-up growth will occur - only 18% of growth remains after entry into puberty.
2.3 Diagnosis
When evaluating a child with short stature, as with any other endocrine diagnosis, the paediatric endocrinologist often integrates multiple clinical and laboratory data rather than relying solely on results from one particular biochemical assay.
Evaluation for GH deficiency in a child who is short (i.e. whose height is more than 2 standard deviations (SD) below the mean expected value) should not be initiated until other causes of growth failure (such as hypothyroidism, chronic systemic disease, Turner syndrome or skeletal disorders) have been excluded. A comprehensive clinical and auxological assessment, combined with a bone age X-ray and biochemical tests of the GH-insulin-like growth factor (IGF) axis, is then warranted. Auxological assessment primarily comprises careful evaluation of a child's growth rate and velocity, together with assessment of the family history of growth and height patterns.
3. Currently accepted, evidence-based indications for GH
3.1 GH deficiency
3.1.1 Clinical considerations
GH deficiency (Fig. 5) encompasses numerous abnormalities within the GH-IGF axis. Poor growth can result from deficiency of GH, the GH molecule being biologically inert, GH receptor problems or downstream defects in the GH signal-transduction pathway, abnormalities of IGF generation, or end-organ unresponsiveness. GH and IGF-1 are ultimately responsible for skeletal growth.

While poor growth rate or short stature may be the cardinal clinical feature of GH deficiency, the following characteristics may also suggest the diagnosis:
• Cherubic faces, truncal obesity or small hands and feet
• Neonates with hypoglycaemia, prolonged jaundice or microphallus (see below)
• Traumatic delivery or breech presentation
• Cranial irradiation or chemotherapy
• Head trauma or central nervous system infection
• Craniofacial midline abnormalities
• Consanguinity.
Because GH is normally secreted in a pulsatile manner, with very low levels during the day and six or seven spontaneous spikes most commonly occurring during deep sleep, clinicians cannot rely on a random blood sample for measurement of GH levels. Instead, endocrinologists can stimulate the pituitary gland pharmacologically (e.g. with insulin, clonidine, or L-dopa) or physiologically (via exercise or sleep) to secrete GH and observe the response. However, such testing relies on an arbitrary definition of what constitutes a normal or a subnormal response; the assays used to measure the response are not standardised and have poor reproducibility; the response to testing is age dependent; and there is overlap between responses in people with and without GH deficiency. Furthermore, the tests are uncomfortable, expensive, and at times involve an element of risk to the patient. Consequently, GH-stimulation testing should be performed in a supervised and experienced unit, and children in whom it is to be performed should be referred to a paediatric endocrinologist.
To improve diagnostic accuracy, in addition to GH measurements, consideration has been given to assessing GH-dependent components of the IGF system, such as IGF-1 and IGFBP-3. Although IGFBP-3 testing is not available in South Africa, IGF-1 testing can be performed. However, evaluation of IGF also has limitations. Analysis of IGF-1 levels is far from clear-cut; results are markedly affected by a child's age and are also nutrition-dependent. Moreover, comparisons between results of IGF-1 assays and GH provocative tests are far from 100% concordant. It is likely that clinicians are dealing with a continuum of GH-IGF secretion, and clear discrimination between GH deficiency and borderline GH-IGF production may not be possible. It is probably unrealistic to expect - or ever to have expected - that a biological process as complex as growth can be reduced to one or two simple diagnostic tests.
At this time, the diagnosis of GH deficiency in children with short stature may need to be based on auxology and supported by either provocative GH testing or careful evaluation of the IGF system. This approach may result in some over-diagnosis and over-treatment, but this is preferable to failing to diagnose or treat appropriate children during a window of time that will allow them to grow.3
3.1.2 Anabolic and metabolic indications for the treatment of GH deficiency in children
In addition to reduced linear growth, children with severe GH deficiency demonstrate reduced lean body mass, increased body fat, subnormal bone density, and a tendency to develop lipid abnormalities. Importantly, in addition to promoting linear growth, GH has considerable and favourable physiological effects on body composition through its actions on adipose tissue, muscle accretion and bone metabolism. GH directly stimulates osteoblast and osteoclast differentiation, and promotes the accretion of bone mass during childhood and adolescence.
Children with GH deficiency should be re-tested once growth is complete, and the transition should be made to adult GH replacement therapy should this be required. Patients with multiple pituitary hormone deficiencies may not require retesting as the greater the number of pituitary hormone deficits, the greater the likelihood of permanent GH deficiency.
3.1.3 Height outcomes, GH dose and follow-up
Before 1985, the adult height of untreated children who had GH deficiency was around 142 cm. Those treated before the introduction of synthetic GH in 1985 were treated with cadaveric GH. The results of GH treatment were attenuated by its limited supply and variable biological potency. Since 1985, when recombinant GH became available, the adult height of children treated with this product has ranged from 165 to 172 cm. The marked improvement in adult height has been attributed to higher doses of recombinant GH (0.2 - 0.3 mg/kg/week), chronological and bone age at the start of treatment, longer duration of therapy, correction of height deficit before the onset of puberty, frequency of hormone administration (daily administration is superior to all other frequencies), increased parental heights, and compliance with therapy. The usual starting dose of GH is between 0.2 and 0.3 mg/kg/week. Regular follow-up visits are required to document ongoing response, enabling the detection and subsequent investigation of any suboptimal growth responses.
3.1.4 GH deficiency in the neonatal period
Clinical associations of GH deficiency in the neonatal period include breech presentation, midline facial defects, microphallus, hypoglycaemia and prolonged jaundice. A low GH level (<20 ng/ml) in the presence of hypoglycaemia confirms GH deficiency. However, this measurement is not always easy to obtain, and routine GH testing, including evaluation of IGF-1 levels, is not easy to perform or to interpret. Prevention of hypoglycaemia, which may cause damage to the developing brain, is a primary concern and GH is effective in this regard. Replacement of other pituitary hormones, should they too be deficient, is also essential.
3.2 Turner syndrome
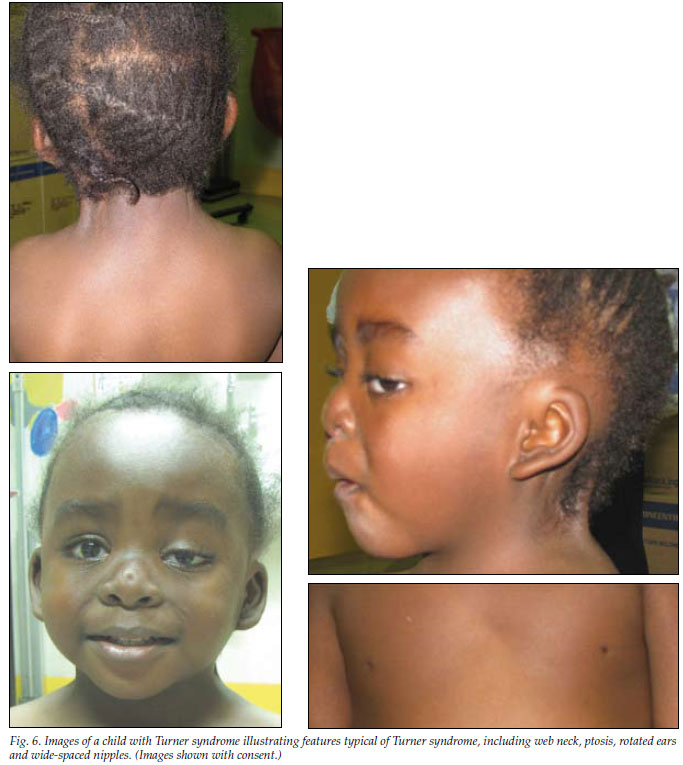
Turner syndrome (Fig. 6) is a common chromosomal abnormality (45,X0 karyotype and variants), occurring once in every 2 000 - 5 000 live-born phenotypic females. The cardinal clinical features are short stature and primary gonadal failure, both of which are apparent in virtually all patients. However, these are not the only features, and comprehensive multidisciplinary care is required to investigate and manage other components of the syndrome. Other typical features include web neck, lymphoedema, ptosis, strabismus, low posterior hairline, high-arched palate, simple rotated ears, recurrent otitis media, wide-spaced nipples, scoliosis and kyphosis, cubitus valgus, concave nails, and short 4th and 5th metacarpals. Abnormalities associated with Turner syndrome involve several major body systems, including the cardiovascular, renal, auditory and endocrine (particularly influencing growth, gonadal failure, thyroid disorders and coeliac disease) systems. Psychosocial wellbeing is also likely to be affected.
The average height of untreated women with Turner syndrome is 142 cm (20 cm less than the average adult female). Growth in such individuals is subnormal in infancy and a further 12 cm is lost by puberty, which is often delayed or absent and associated with an attenuated growth spurt when it does occur. Because growth velocity declines from an early stage, many affected girls are significantly below the 5th percentile for height by the age of 2, 3 or 4 years, thus permitting early diagnosis and intervention.
Patients with Turner syndrome are not GH deficient, and GH stimulation testing is therefore not generally needed (although it may be performed when individuals are not growing in accordance with the Turner-specific growth chart). In 1995, GH was approved in the USA for the treatment of short stature associated with Turner syndrome; published data from a prospective, randomised clinical study performed at multiple sites in the USA have shown GH therapy to have a beneficial effect on adult height in patients with Turner syndrome.4 Recently published results of an extensive randomised controlled trial of GH performed in Canada using a dose of 0.3 mg/kg/week showed that patients treated with GH attained a mean final height of 149 cm compared with 142 cm in those who remained untreated; most benefit was obtained in patients treated for the longest time.5 Indeed, available evidence highlights the need for early initiation of GH treatment,6 and administration of GH from 9 months of age should be considered.
A major issue in the treatment of Turner syndrome is determining when to introduce oestrogen therapy. Past practices that significantly delayed the introduction of oestrogen to maximise height attainment are no longer favoured. Early GH treatment may allow girls to reach near-normal heights by pubertal age, facilitating bone accretion and enabling appropriately timed pubertal induction and normal psychosocial adaptation. During pubertal induction, transdermal oestrogen replacement may confer advantages over oral replacement therapy in terms of final height.7
Appropriate clinical management of Turner syndrome therefore involves early administration of GH, enabling patients to reach a more acceptable adult height sooner, and permitting the introduction of oestrogen at 12 - 13 years of age, if height deficit is no longer substantial.
3.3 Prader-Willi syndrome
The value of GH therapy in correcting abnormalities that are discrete from the issue of height is well illustrated by Prader-Willi syndrome. This is an inherited disorder of chromosome 15, affecting 1 in 15 000 children. It is characterised by early failure to thrive, low muscle tone, poor feeding, neurodevelopmental delay and cognitive deficits. In addition, there is growth failure with adult short stature, osteoporosis, hypogonadism, hyperphagia and excessive body fat,8 with the development of morbid obesity and all of its negative health consequences.
In 2000, GH was registered for the treatment of paediatric Prader-Willi syndrome patients with growth failure, for improvement of growth and body composition. Numerous beneficial effects of GH include improvements in growth, physical appearance, functional muscle mass and infant neurodevelopment.9-13
From an ethical viewpoint, responsiveness to GH therapy and the improved functional outcome should determine entitlement to treatment, not a diagnosis of GH deficiency, particularly in patients treated at a young age. The labelled recommended dose of GH is 0.24 mg/kg/week, but in clinical practice improvements in a range of parameters are seen at lower doses.
3.4 SGA children with failure to show catch-up growth
Approximately 3% of children are born SGA, defined as a birth length and/or weight >2 SD below the mean for sex and age. Most of these children will have sufficient post-natal catchup growth to normalise their stature by the age of 2 years.14 However, in around 10% of SGA individuals, body size will remain small throughout childhood; around 20% of short adults were born SGA.15
GH therapy has been proven to be safe and effective in reducing the adult height deficit that may otherwise be apparent in short SGA children. For SGA children who are not extremely short (-2.0 to -3.0 SD), current data support a GH dose of 0.23 mg/kg/week from starting therapy until adult height is attained, particularly if treatment is initiated at a young age. For shorter children, whose height is below -3.0 SD, benefit may be obtained from an approach whereby catch-up growth is achieved with a GH dose of 0.35 - 0.4 mg/kg/week; a GH dose of 0.23 mg/kg/week can then be given to ensure long-term growth through to adult height. In practice, this can be achieved by initiating GH at a dose of 0.35 mg/kg/week and leaving the dose unchanged; as height and weight increase, the dose will automatically taper to 0.23 mg/kg/week.16
3.5 Idiopathic short stature
Idiopathic short stature syndrome is defined as significantly short stature (at least -2.5 SD) and a persistently low growth rate in the absence of any evidence of systemic disease, malnutrition, hypothyroidism, chromosomal abnormality, or classic GH deficiency on provocative testing. There are multiple causes, including genetic or familial short stature, constitutional delay of puberty, dysfunction affecting the GH axis (notably IGF deficiency) or partial GH insensitivity syndrome, combinations of constitutional delay and genetic short stature, and causes of unknown origin. As a group, children with idiopathic short stature are as short as those with GH deficiency, and they grow at a similarly slow rate.17
A recent Cochrane review concluded that there is some evidence that recombinant human GH improves short-term growth and (near) final adult height in children with idiopathic short stature.18 Results from a total of 741 children reported in 10 studies of between 6 months' and 6.2 years' duration showed that individuals treated with GH remained relatively short when compared with peers of normal stature. However, in one study girls treated with GH have been reported to have attained a mean height 7.5 cm greater than untreated controls (155.3 v. 147.8 cm),19 and others have found a mean height 3.7 cm greater for GH-treated children compared with placebo-treated counterparts.20 Further data on final height are awaited to clarify the use of GH for correcting idiopathic short stature.
GH treatment could be considered for children with idiopathic short stature who exhibit a significant height deficiency (-2.5 SD (instead of -2.0 SD) compared with that of appropriate controls), a persistently low growth rate (less than the 25th percentile when observed over a period of at least 1 year, or less than the 40th percentile over 2 years), and no evidence of systemic disease.3
Cost is one of the greatest concerns relating to the treatment of idiopathic short stature. Given the variable, and sometimes unknown, causes of the condition, it is to be expected that while some patients will respond well to GH therapy, for others the treatment may be less effective. Intention-to-treat analyses in clinical trials probably underestimate the benefits of therapy for some patients, as those who respond poorly may withdraw from therapy but are still analysed in the treatment arms. This is good clinical practice in the research setting, but does not take into consideration the fact that patients who demonstrate a favourable clinical response are more likely to continue with treatment than those in whom the response is poor - a scenario which is more akin to day-to-day clinical practice. Given such behaviour, patients with a poor response to treatment may be withdrawn from ineffective, expensive therapy. Targeted treatment of those children with idiopathic short stature who have the greatest potential for growth is critical to improve the cost-effectiveness of GH therapy.21
PAEDS-SA recommends that children with idiopathic short stature be offered a trial of GH therapy for a period of 12 months, during which time significant increases in annualised height velocity would be expected in those individuals who are likely to benefit.22 If effective, therapy should be continued for as long as clinical benefit - defined as ongoing catch-up growth or maintenance of a normal growth velocity - is apparent. GH should be discontinued once a height at or above the 3rd percentile of the adult height chart is achieved, if annualised growth velocity declines below 3 cm/year, or if growth plates close before this percentile is attained.
3.6 Chronic renal insufficiency
Chronic renal insufficiency is characterised by an insidious and irreversible loss of nephron function and glomerular filtration. This progresses to chronic renal failure and ultimately to end-stage renal disease, which necessitates dialysis and/or renal transplantation. There are no accurate data on the prevalence of chronic renal insufficiency in the paediatric population.
Growth failure is one of the most common clinical features of chronic renal insufficiency, and stunted growth is already apparent in many children at the time of presentation. It remains a major obstacle to satisfactory psychosocial rehabilitation. Most children affected by chronic renal insufficiency fail to attain their genetic height potential (with a mean adult height 2.0 SD below the mean for unaffected individuals) because of a combination of factors including poor growth during critical rapid growth periods such as the first 2 years of life and puberty, delayed, abbreviated puberty (with an attenuated growth rate), malnutrition, acidosis, and renal osteodystrophy. If children with chronic renal insufficiency are to be treated with GH, their nutritional and medical status must be optimised to ensure an adequate response to therapy.
GH was approved by the United States Food and Drug Administration for management of growth before renal transplantation in individuals with chronic renal insufficiency. Benefits of GH therapy have been documented for up to 5 years following enrolment of a large group of children with chronic renal failure into a multicentre randomised, double-blind, placebo-controlled study.23 At the end of the first year of this study, the growth rate was 6.5 cm/year in control patients compared with 10.7 cm/year in patients who received GH.24 At the end of the second year, growth rates in the two groups were 5.5 and 7.8 cm/year, respectively. The average increase in relative height in the GH-treated patients increased from -2.94 SDS to -1.55 SDS, compared with a further decline from baseline in the untreated patients after the first 2 years. When therapy was changed from placebo to GH there was a dramatic improvement in height SD scores, from a pre-therapy score of -2.9 SD to -1.9 SD. GH treatment did not accelerate bone age or progression to end-stage renal disease. Although it had been thought that correcting renal status by dialysis or transplantation would normalise growth in patients with chronic renal insufficiency, results from studies assessing these treatments have been disappointing.25 Current evidence supports the early introduction of GH in patients with deteriorating renal function to preserve normal growth - catchup growth is poor during dialysis and very limited after renal transplant.
4. Safety concerns
4.1 General overview
Extensive data collected from large numbers of children and adults treated with GH indicate that, for current approved indications, GH is safe.26 However, it is appropriate to briefly consider several issues in relation to safety.
4.2 Tumour occurrence or recurrence
Evidence from published literature does not support the existence of a relationship between tumour recurrence and GH therapy.26-30 However, GH is contraindicated in children with an active malignant condition. If GH deficiency is due to an intracranial tumour, absence of growth or recurrence should be documented for 6 - 12 months before initiation of GH therapy. Considering the limitations of published studies, continued surveillance is suggested in children or adults who have a history of tumours and are receiving treatment with GH.
4.3 Intracranial hypertension
In an international database, the incidence of intracranial hypertension was 1.1/1 000 cases in the first year of GH treatment, with a bias towards patients with chronic renal failure or Turner syndrome.31 A national database in Australia and New Zealand reported the overall incidence of intracranial hypertension in children receiving GH therapy to be 1.2/1 000 cases.32 Patients with GH deficiency were most commonly affected, with an incidence of 6.5/1 000 cases.32
4.4 Other side-effects
Other minor side-effects of GH treatment include breast tenderness and mild degrees of gynaecomastia, water retention, an increased number of cutaneous nevi (without malignant potential), and slipped capital femoral epiphysis. These side-effects are not life-threatening, and in all instances appropriate management is available.
5. Deciding coverage for GH treatment
Mutual understanding among payers (in both the private and state sectors) and providers regarding funding for GH treatment has never been more urgently needed. The formation of a GH review committee chaired by a paediatric endocrinologist and consisting of a medical director, case manager, pharmacy director and administrative assistant/ analyst would aid the evaluation of applications for GH use on a case-by-case basis when criteria for therapy are not clear. The respective parties have a duty to supply the committee with all information relevant to the decision-making process. This should include pertinent information relating to the patient (age, sex, pubertal status, parental heights (if available), etc.), together with a brief history and the suspected diagnosis; auxological data (preferably a growth chart) with more than 6 months' data; results of laboratory studies, including, but not limited to, GH-stimulation and IGF-1 test results; and a bone age report, or preferably an original X-ray. After initiation of GH therapy, a rolling review process, performed every 6 months, should determine whether treatment endpoints are being achieved and the therapy is appropriate.
6. Proviso
The PAEDS-SA recommendations are based on sound scientific evidence and the collective clinical experience of the group.
PAEDS-SA recognises the financial limitations that exist in the state sector and recommends that GH use is limited to tertiary-level hospitals managing individuals with short stature, with patients being treated by paediatric endocrinologists or paediatricians in consultation with regional paediatric endocrinologists.
We continue to advocate GH use for neonatal GH deficiency with hypoglycaemia as an absolute indication if life expectancy is normal and there are no major congenital malformations or syndromes that would limit the benefit derived from GH therapy.
We also advocate the use of GH for managing short stature in children with proven GH deficiency, whatever the cause, where there is expected benefit based on age at presentation and growth plate potential.
Side-effects associated with GH use are rare, and the anticipated benefits of treatment almost always outweigh the potential risks.
7. Addendum
In South Africa, the commercially available GH products are deemed to be equivalent in their growth-promoting ability. When GH treatment is initiated, the choice of which product to use may be influenced by the payer in the light of financial considerations. However, when patients have already started therapy, the product selection should be directed by the attending doctor.
Payers should allocate four visits per year for each patient to receive consultation with an endocrine specialist for documentation and monitoring of ongoing care.
8. Conflict of interest disclosure
Dr David Segal has no conflict of interest disclosure to make. Editing assistance was provided by Bioscript Stirling Ltd, UK, and funded, through an unrestricted educational grant, by Novo Nordisk South Africa.
9. References
1. Wright CM, Cheetham TD. The strengths and limitations of parental heights as a predictor of attained height. Arch Dis Child 1999; 81: 257-260. [ Links ]
2. Tanner JM, Davies PS. Clinical longitudinal standards for height and height velocity for North American children. J Pediatr 1985; 107: 317-329. [ Links ]
3. Rosenfeld R, Allen DB, MacGillivray MH, et al. Growth hormone use in pediatric growth hormone deficiency and other pediatric growth disorders. Am J Manag Care 2000; 6: 15 suppl, S805-S816. [ Links ] [ Links ]
5. Stephure DK; Canadian Growth Hormone Advisory Committee. Impact of growth hormone supplementation on adult height in Turner syndrome: results of the Canadian randomized controlled trial. J Clin Endocrinol Metab 2005; 90: 3360-3366. [ Links ]
6. Soriano-Guillen L, Coste J, Ecosse, E, et al. Adult height and pubertal growth in Turner syndrome after treatment with recombinant growth hormone. J Clin Endocrinol Metab 2005; 90: 5197-5204. [ Links ]
7. Ankarberg-Lindgren C, Elfving M, Wikland KA, Norjavaara E. Nocturnal application of transdermal estradiol patches produces levels of estradiol that mimic those seen at the onset of spontaneous puberty in girls. J Clin Endocrinol Metab 2001; 86: 3039-3044. [ Links ] [ Links ]
9. Carrel AL, Lee PDK, Mogul HR. Growth hormone and Prader-Willi syndrome. In: Butler MG, Lee PDK, Whitman BY, eds. Management of Prader-Willi Syndrome. 3rd ed. New York: Springer, 2006: 201-241. [ Links ]
10. Eiholzer U. Prader-Willi Syndrome: Effects of Human Growth Hormone Treatment. Zurich: Karger, 2001. [ Links ]
11. Eiholzer U, Lee PDK. Medical considerations in Prader-Willi syndrome. In: Butler MG, Lee PDK, Whitman BY, eds. Management of Prader-Willi Syndrome. 3rd ed. New York: Springer, 2006: 97-152. [ Links ]
12. Lee PD. Disease management of Prader-Willi syndrome. Expert Opin Pharmacother 2002; 3: 1451-1459. [ Links ]
13. Lindgren AC, Hagenäs L, Müller J, et al. Growth hormone treatment of children with Prader-Willi syndrome affects linear growth and body composition favourably. Acta Pediatr 1998; 87: 28-31. [ Links ]
14. Hokken-Koelega AC, De Ridder MA, Lemmen RJ, et al. Children born small for gestational age: do they catch up? Pediatr Res 1995; 38: 267-271. [ Links ]
15. Karlberg J, Albertsson-Wikland K. Growth in full-term small-for-gestational age infants: from birth to final height. Pediatr Res 1995; 38: 733-739. [ Links ]
16. de Zegher F, Hokken-Koelega A. Growth hormone therapy for children born small for gestational age: height gain is less dose dependent over the long term than over the short term. Pediatrics 2005; 115: e458-e462. [ Links ]
17. Wit JM, Rekers-Mombarg LT, Cutler GB, et al. Growth hormone (GH) treatment to final height in children with idiopathic short stature: evidence for a dose effect. J Pediatr 2005; 146: 45-53. [ Links ]
18. Bryant J, Baxter L, Cave CB, Milne R. Recombinant growth hormone for idiopathic short stature in children and adolescents. Cochrane Database Syst Rev 2007; (3): CD004440. [ Links ]
19. McCaughey ES, Mulligan J, Voss LD, Betts PR. Randomised trial of growth hormone in short normal girls. Lancet 1998; 351: 940-944. [ Links ]
20. Leschek EW, Rose SR, Yanovski JA, et al. Effect of growth hormone treatment on adult height in peripubertal children with idiopathic short stature: a randomized, double-blind, placebo-controlled trial. J Clin Endocrinol Metab 2004; 89: 3140-3148. [ Links ]
21. Lee JM, Davis MM, Clark SJ, Hofer TP, Kemper AR. Estimated cost-effectiveness of growth hormone therapy for idiopathic short stature. Arch Pediatr Adolesc Med 2006; 160: 263-269. [ Links ]
22. Bakker B, Frane J, Anhalt H, Lippe B, Rosenfeld RG. Height velocity targets from the national cooperative growth study for first-year growth hormone responses in short children. J Clin Endocrinol Metab 2008; 93: 352-357. [ Links ]
23. Fine RN, Kohaut E, Brown D, Kuntze J, Attie KM. Long-term treatment of growth retarded children with chronic renal insufficiency, with recombinant human growth hormone. Kidney Int 1996; 49; 781-785. [ Links ]
24. Fine RN, Kohaut EC, Brown D, Perlman AJ. Growth after recombinant human growth hormone treatment in children with chronic renal failure: report of a multicenter randomized double-blind placebo-controlled study. Genentech Cooperative Study Group. J Pediatr 1994; 124: 374-382. [ Links ]
25. von Lilien T, Gilli G, Salusky IB. Growth in children undergoing continuous ambulatory or cycling peritoneal dialysis. In: Schärer K, ed. Paediatric and Adolescent Endocrinology. Vol 20. Basel: Karger, 1989: 27-35. [ Links ] [ Links ]
27. Blethen SL, Allen DB, Graves D, August G, Moshang T, Rosenfeld R. Safety of recombinant deoxyribonucleic acid-derived growth hormone: The National Cooperative Growth Study experience. J Clin Endocrinol Metab 1996; 81: 1704-1710. [ Links ]
28. Moshang T Jr, Rundle AC, Graves DA, Nickas J, Johanson A, Meadows A. Brain tumor recurrence in children treated with growth hormone: the National Cooperative Growth Study experience. J Pediatr 1996; 128: S4-S7. [ Links ]
29. Ogilvy-Stuart AL, Ryder WD, Gattamaneni HR, Clayton PE, Shalet SM. Growth hormone and tumour recurrence. BMJ 1992; 304: 1601-1605. [ Links ]
30. Ogilvy-Stuart AL, Shalet SM. Tumour occurrence and recurrence. Horm Res 1992; 38: suppl 1, 50-55. [ Links ]
31. Ranke MB, Wilton P, eds. Growth Hormone Therapy in KIGS-10 Years' Experience. Heidelberg, Leipzig: Barth, 1999: 349-364 [ Links ]
32. Crock PA, McKenzie JD, Nicoll AM, et al. Benign intracranial hypertension and recombinant growth hormone therapy in Australia and New Zealand. Acta Paediatr 1998; 87: 381-386. [ Links ]
On behalf of the Paediatric and Adolescent Endocrine and Diabetes Society of South Africa
Correspondence to: Dr David Segal, Centre for Diabetes and Endocrinology, 18 Eton Road, Parktown, Johannesburg, 2198, tel. 011 726-0016, fax 086 6890700, e-mail david.segal@wits.ac.za


{kind=link}