










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSAMJ: South African Medical Journal
On-line version ISSN 2078-5135
Print version ISSN 0256-9574
SAMJ, S. Afr. med. j. vol.99 n.1 Pretoria Jan. 2009
SCIENTIFIC LETTER
Hereditary C1 esterase deficiency in a Zulu kindred
E MoranI; G S IsaacsII; B NaidooII; D J PudifinIII
IMB BCh, PhD, MRCP. John Radcliffe Hospital, Oxford, UK; and formerly of Hlabisa Hospital, Hlabisa, KwaZulu-Natal
IIMB ChB. Department of Medicine, King Edward VIII Hospital, Durban
IIIMB ChB, FCP (SA), FRCP. Nelson Mandela School of Medicine, University of KwaZulu-Natal, Durban
To the Editor: Hereditary angio-oedema is a rare genetic disorder characterised by recurrent episodes of potentially life-threatening angio-oedema. Despite the apparent absence of variation in incidence among ethnic groups, this is the first reported series in a sub-Saharan African family, and it is likely that the disease remains largely undiagnosed in southern Africa. Although cost and availability limit the use of certain treatments which are available in resource-rich health systems, early diagnosis allows patient education and the initiation of less costly, more widely available prophylactic medications that are life-saving in individuals at risk of severe attacks. The diagnosis should be considered in the differential diagnosis of those presenting with apparent allergies and drug reactions.
Case description and diagnosis
Several family members who had all experienced repeated episodes of angio-oedema were seen at Hlabisa District Hospital and King Edward VIII Hospital. The first (NM1), a 16-year-old girl, presented twice to Hlabisa with acute onset facial swelling and respiratory difficulty. The swelling of her face and tongue was unresponsive to intramuscular adrenaline and intravenous steroids. On both occasions, she developed stridor and required endotracheal intubations when severe laryngeal oedema was noted. The oedema settled spontaneously over 2 -3 days. She had had similar, milder episodes about twice a year over the previous 10 years, and other family members had similar episodes.
The second patient (NM2), a 14-year-old cousin of NM1, was well at the time of assessment. She gave a history of attacks of swelling of the lips, face and neck which had started at a young age and resolved spontaneously. She said her brother experienced similar attacks and that her father had died as a result of a severe episode.
NM3, the 12-year-old brother of NM1, presented to Hlabisa with spontaneous facial swelling which subsided after the administration of freeze-dried plasma. Similar episodes had occurred about twice a year over the previous 5 years.
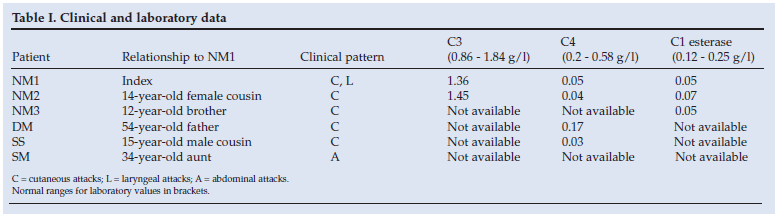
There were no identifiable precipitating factors for the angio-oedema in the 3 patients, and they were otherwise well and physically normal. Since circumstances strongly suggested the diagnosis of familial angio-oedema, complement function was investigated, which confirmed a low C4 level and deficiency of C1 esterase in all 3.
Three other family members (SS, DM and SM) were seen only once as outpatients, and limited data are available (Table I). SS and DM had experienced mild cutaneous episodes. SM, the 34-year-old aunt of NM1, had experienced recurrent episodes of abdominal pain, sometimes associated with facial swelling. A laparotomy and cholecystectomy had been performed following ultrasound demonstration of a single gallstone, but episodes of pain persisted. There were no abnormal signs apart from mild generalised abdominal tenderness during the attacks, which always settled spontaneously over a few days. Full blood count, serum urea, electrolytes and liver function were normal when the patient was seen.
These findings indicate that this kindred of African/Zulu descent harbours one of the mutations giving rise to deficiency of C1 esterase, and this is believed to be the first report of this abnormality in this ethnic group. (The literature search was performed using Pubmed, and the terms Africa and African and Zulu in combination with each of C1 esterase inhibitor, hereditary angio-oedema and hereditary angioedema. Although cases have been identified in North Africa and North America among individuals described as 'African-American', there were no cases reported among those of confirmed sole sub-Saharan African descent.)
Pathogenesis and clinical features of hereditary angio-oedema
Hereditary angio-oedema (HA) is a rare autosomal dominant disorder characterised by recurrent episodes of angio-oedema without urticaria or pruritus. Incidence is estimated at around 1/50 000, with no described variation between ethnic groups. Most commonly affecting the skin and tissues of the upper airways, attacks are self-limited but laryngeal involvement can lead to airway obstruction and death. It is a consequence of either a deficiency (type 1 - 85% of affected families) or a dysfunction (type 2) of C1 inhibitor (C1-INH) - a protein that prevents complement activation by both the classic route and the mannan-binding pathway.1 C1-INH is additionally involved in the regulation of the coagulation and fibrinolytic and kinin-generating pathways and, although the specific mediator responsible for the generation of angio-oedema has not been identified, increasing evidence implicates the vasodilatory protein bradykinin. The gene for C1-INH is located on chromosome 11, and well over 100 mutations have been identified in HA patients, most of which are single base-pair mutations. Around a quarter of cases are due to new mutations. The severity of clinical attacks varies even within the same family possessing the same mutation; the factors influencing severity are not known. Some individuals found to have C1-INH deficiency through family screening never develop attacks.1
Although attacks of angio-oedema may occur spontaneously with no obvious precipitant, some patients may be able to identify specific triggers such as dental work, mild trauma, stress, excitement, certain foods, oestrogen, menstruation, and angiotensin-converting enzyme (ACE) inhibitors. A prodrome of mood or skin changes may precede an episode. Attacks may be cutaneous, abdominal or laryngeal1 and, although often affecting a single site, may affect two or more sites (e.g. facial swelling progressing to laryngeal oedema, as in NM1). Half of affected individuals experience all three forms of attack in their lifetimes; 40% experience their first attack before age 5 years, and 75% by age 15 years. Repeated attacks are uncommon before adolescence, after which attack frequency increases.
Cutaneous attacks tend to affect the hands, feet, face and genitals, with swelling lasting 2 -4 days. Abdominal attacks cause pain, nausea, vomiting and diarrhoea (because of bowel wall oedema). Most patients experience an abdominal attack at some point in their lives that lasts around 4 days, with symptoms peaking on day 2. Resemblance to the acute surgical abdomen leads to unnecessary surgery in around a third of patients with undiagnosed HA (as in SM). Laryngeal attacks occur in half of all patients at some point in their lives, and are less common after the age of 45 years. They can occur in the absence of swelling of the upper airway and lips. It usually takes a few hours to progress from first symptom to potential airway obstruction, but obstruction can occur within half an hour of first symptom.
Diagnosis and treatment
Initial screening is by measuring C4 and C1 inhibitor levels in a patient with a suggestive personal or family history (e.g. repeated episodes of facial swelling or abdominal pain or an affected relative). Both levels are likely to be reduced in an affected individual; 90% of HA patients have low C4 levels even when asymptomatic, and most have low C4 levels during attacks. A normal C4 in an asymptomatic patient does not exclude HA.2 Confirmation is by full complement studies, ideally performed on two samples taken at least 4 weeks apart. Patients should be off all therapy for HA for at least 3 weeks before testing. If C1q is normal and C4 and C1-INH levels are low, the diagnosis may be HA type 1 (deficient C1-INH). If C1q is normal and C4 is low, but C1-INH levels are normal or raised, the diagnosis may be HA type 2 (dysfunctional C1-INH) and functional assays of C1-INH should be performed. If function is less than 30% of normal, the diagnosis is probably HA type 2. Genetic testing is not required to confirm the diagnosis in adults. In children <1 year of age, complement levels are difficult to interpret, and the diagnosis should be confirmed by either repeating the assays when the child is older or by genetic testing.
If a patient with possible HA has no family history or is older than 40 years at first presentation, a rare form of acquired C1-INH deficiency is possible. The diagnosis is suggested by low C1q and C4 levels. Such patients should be screened for malignancy, as 85% are found to have lymphoproliferative disorders. Genetic studies may be required. In addition to acquired C1-INH deficiency, the differential diagnosis for HA includes autoimmune disease (e.g. systemic lupus erythematosus, which may cause an isolated low C4), allergy, anaphylaxis, idiopathic or drug-induced angio-oedema, and parasitic infection.
Treatment of acute attacks
Swift emergency management of laryngeal attacks can be life saving; airway management is paramount, and severely affected individuals may require urgent intubation or cricothyroidotomy. Antihistamines, steroids and adrenaline are not effective but are often given, particularly if the diagnosis is in doubt or not confirmed. The most effective medical treatment for acute episodes of HA is intravenous C1 esterase inhibitor protein,3 which is manufactured from pooled human plasma, and where available is considered the treatment of choice for laryngeal and severe abdominal attacks. However, it is expensive and not widely used outside Europe, Canada and certain other countries. Fresh-frozen plasma contains several complement proteins and is considered first-line therapy where C1 esterase inhibitor protein is not available.4 These therapies carry the risk of transmission of blood-borne viruses, and trials of both recombinant and treated C1 esterase inhibitor preparations are under way.5 Tranexamic acid is used in the treatment of laryngeal, abdominal and severe cutaneous attacks.1,6 It is most effective if given early in the course of symptoms; its mechanism of action is unclear. Symptomatic therapy for pain, vomiting and abdominal cramps should also be given.
Prophylaxis of attacks
Synthetic androgens such as danazol are effective in short- and long-term prophylaxis by increasing hepatic production of C1-INH, but have significant side-effects.7 Long-term prophylaxis should be considered if patients have experienced laryngeal attacks (particularly if C1-INH protein replacement is not available) or more than one severe attack a month. To avoid side-effects, the dose should be titrated to find the lowest effective dose. Short-term prophylaxis should be given to patients undergoing elective surgery and may also be indicated before dental work. Although short-term treatment with synthetic androgens is usually well tolerated by children and adults, long-term treatment can lead to menstrual irregularities in premenopausal women, and weight gain in men and women. Long-term use should be avoided in children and pregnant women, who should instead receive tranexamic acid which, although less effective, can be used for prolonged periods. C1 esterase inhibitor protein can be used for long-term prophylaxis in selected patients.3
Non-medical measures important in the long-term management of HA include education and avoidance of any identified trigger. Patients should be counselled to seek early hospital care, particularly if they have a record of laryngeal attacks, and be provided with an emergency treatment plan to show to hospital emergency departments to facilitate prompt effective therapy.
References
1. Gompels MM, Lock RJ, Abinun M, et al. C1 inhibitor deficiency: consensus document. Clin Exp Immunol 2005; 139(3): 379-394. [ Links ]
2. Karim Y, Griffiths H, Deacock S. Normal complement C4 values do not exclude hereditary angioedema. J Clin Pathol 2004; 57(2): 213-214. [ Links ]
3. Cicardi M, Zingale LC, Zanichelli A, Deliliers DL, Caccia S. The use of plasma-derived C1 inhibitor in the treatment of hereditary angioedema. Expert Opin Pharmacother 2007; 8(18): 3173-3181. [ Links ]
4. Prematta M, Gibbs JG, Pratt EL, Stoughton TR, Craig TJ. Fresh frozen plasma for the treatment of hereditary angioedema. Ann Allergy Asthma Immunol 2007; 98(4): 383-388. [ Links ]
5. Davis AE 3rd. New treatments addressing the pathophysiology of hereditary angioedema. Clin Mol Allergy 2008; 6: 2. [ Links ]
6. Sheffer AL, Austen KF, Rosen FS. Tranexamic acid therapy in hereditary angioneurotic edema. N Engl J Med 1972; 287(9): 452-454. [ Links ]
7. Bork K, Bygum A, Hardt J. Benefits and risks of danazol in hereditary angioedema: a long-term survey of 118 patients. Ann Allergy Asthma Immunol 2008; 100(2): 153-161. [ Links ]
Accepted 21 July 2008.
Corresponding author: E Moran (ed@moran.org)


{kind=link}