










Servicios Personalizados
Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkSAMJ: South African Medical Journal
versión On-line ISSN 2078-5135
versión impresa ISSN 0256-9574
SAMJ, S. Afr. med. j. vol.98 no.6 Pretoria jun. 2008
SCIENTIFIC LETTERS
Pyruvate kinase deficiency in a South African kindred caused by a 1529A mutation in the PK-LR gene
Pierre M DurandI; Theresa L CoetzerII
IMB BCh, MSc, MMed. Department of Molecular Medicine and Haematology, University of the Witwa-tersrand and National Health Laboratory Service (NHLS), Johannesburg
IIPhD. Department of Molecular Medicine and Haematology, University of the Witwa-tersrand and National Health Laboratory Service (NHLS), Johannesburg
To the Editor: There is currently no investigation for pyruvate kinase (PK) deficiency in South Africa and nothing is known about local mutations. We describe the implementation of a PK assay and document the first mutation underlying PK deficiency in a South African patient who presented with a haemolytic episode following ARV (antiretroviral) prophylaxis.
PK deficiency is the most common inherited disorder of glycolysis in humans.1 The enzyme catalyses the conversion of phosphoenolpyruvate (PEP) to pyruvate, producing adenosine triphosphate (ATP) from adenosine diphosphate (ADP) in the process. A PK deficiency therefore results in decreased intra-erythrocytic ATP, which cannot be compensated for by oxidative phosphorylation since erythrocytes lack mitochondria. This leads to membrane damage, haemolysis and premature destruction in the spleen.
Epidemiological studies indicated that the PK heterozygote allele frequency ranges from 1% to 5% in Caucasian populations, and one small study suggested the frequency might be twice as high in African-Americans.2 Areas of south-east Asia may have a significantly higher prevalence,2,3 although there have been no large-scale studies conducted in this region. The reason for the relatively high frequency of the abnormal PK allele has been debated. The majority of inherited erythrocyte disorders have been selected by their relative resistance to malaria,4 and we have recently demonstrated that PK deficiency protects against malaria in humans (manuscript accepted for publication in Haematologica, May 2008), which provides a possible explanation.
The prevalence of the disease in South Africa is unknown, and few individual cases have been reported (personal communication, Dr N Alli, Chris Hani Baragwanath Hospital). There is currently no available assay for PK deficiency in South Africa, and nothing is known about the underlying mutations in the PK-LR gene in South Africans and Africans in general. More than 180 mutations cause the disease,3 and it would be interesting to establish the mutation spectrum in southern Africans.
We report the implementation of an assay for PK activity. A patient with suspected PK deficiency was confirmed to have the disease, and the underlying mutation was identified.
Methods
PK assay implementation
The PK assay, based on Beutler's method5 with a few minor modifications, was implemented in the Red Cell Membrane Unit, Department of Molecular Medicine and Haematology at the University of the Witwatersrand National Health Laboratory Service (NHLS) in Johannesburg. Qualitative (screening) and quantitative (confirmatory) assays were implemented, and the screening assay may be added to the routine tests offered by the NHLS. Guidelines for physicians are available from the authors.
The screening and quantitative assays use the same test principle: Plasma and leucocytes are removed from whole blood and the packed erythrocytes resuspended in 0.9% saline. A 1:20 haemolysate is made of the red cells and used to determine PK enzyme activity by the conversion of PEP to pyruvate. This reaction is coupled to a second reaction, which uses lactate dehydrogenase to convert pyruvate to lactate with nicotinamide adenine dinucleotide (reduced form) (NADH) as a co-factor. The oxidation of NADH is determined by a loss of fluorescence (screening assay) or spectrophotometrically at 340 nm (quantitative assay), and is used as a measure of PK activity.
Ethics clearance was obtained from the Human Research Ethics Committee (Medical), of the University of the Witwatersrand to analyse EDTA blood samples from 20 whites and 20 blacks with haematologically normal profiles, to determine a local reference range for PK activity. The following quality assurance issues were addressed: assay precision and reproducibility; assay sensitivity; enzyme stability in whole blood; paediatric samples; and feasibility to implement the assay as a routine test. The assay was validated with blood from a PK-deficient patient.
Mutation analysis
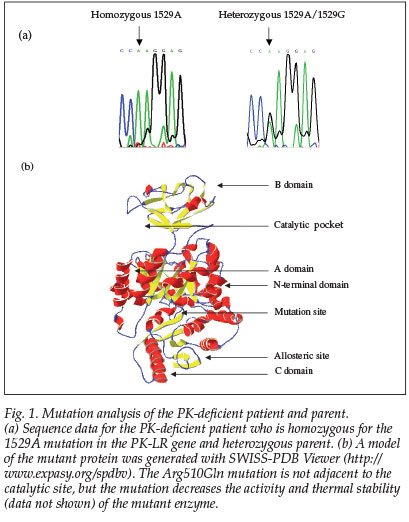
DNA mutation analysis was performed on samples from the PK-deficient patient and 1 parent. In brief, genomic DNA was extracted from the buffy coat6 and exons 8-10, 11 and 12 of the PK-LR gene amplified by polymerase chain reaction (PCR). Automated DNA sequencing was performed by Inqaba Biotech, Johannesburg.
Results and discussion
PK assay implementation
The screening assay detected a decreased PK activity if the sample had an enzyme activity of <50% of the mean. This is considered adequate, since clinically relevant PK deficiency develops when an individual has an enzyme activity of <25% of normal.
There was no difference between the means of the white (13.41 U/g haemoglobin (Hb)) and black (12.64 U/g Hb) groups (p=0.29). The combined mean (± significant deviation (SD)) (N=40) in this study was 13.01±2 U/g Hb and falls within 1 SD of the published reference range.1 The coefficient of variation for the precision and reproducibility of the quantitative assay was 5%. The PK enzyme is stable in EDTA whole blood for 5 days if stored at 4oC, and for 3 days if stored at room temperature. The minimum volume of whole blood required to perform the assay is 0.5 ml, which makes it suitable for samples taken from paediatric patients. The assay was performed on a further 10 samples received from laboratories around the country as part of a 'haemolytic work-up' to demonstrate the applicability and feasibility of the assay in a routine setting.
PK-deficient patient
A 24-year-old white woman with suspected PK deficiency had suffered a haemolytic episode immediately after the prophylactic use of antiretrovirals (3TC, AZT and Crixivan) a year earlier. A referral letter reported haemolytic episodes as an infant but nothing subsequently. The only medication she used was Sibelium for migraines, and she was otherwise well. She was jaundiced and had no clinically evident splenomegaly. At the time of referral, her serum Hb was 9.6g/dl and she had a reticulocytosis (15.6%). The quantitative PK assay confirmed the enzyme deficiency. The patient's mean PK activity was 15% of normal (N=4). One parent was identified as an asymptomatic carrier and had an enzyme activity of 58% of normal (N=3). The second parent was unavailable.
The association between haemolytic crises in PK deficiency and ARVs has not been reported in the literature previously, and this finding is important in South Africa.
Mutation analysis
The patient was homozygous for the 1529A mutation in exon 11 in the PK-LR gene (Fig. 1). This is the most common mutation in northern Europeans and the first mutation described in a South African patient. The parent was heterozygous for the same mutation, which results in an amino acid change from arginine to glutamine at residue 510.
The authors thank the PK-deficient patient and her parent for their co-operation, and Drs I Thompson and P Keene for the referral.
References
1. Eber SW. Disorders of erythrocyte glycolysis and nucleotide metabolism. In: Handin RI, Lux SE and Stossel TP, eds. Blood: Principles and Practice of Haematology. 2nd ed. Philadelphia: Lippincott, Williams & Wilkins, 2003: 1895-1901. [ Links ]
2. Beutler E, Gelbart T. Estimating the prevalence of pyruvate kinase deficiency from the gene frequency in the general white population. Blood 2000; 95: 3585-3588. [ Links ]
3. Zanella A, Fermo E, Bianchi P, Chiarelli LR, Valenti, G. Pyruvate kinase deficiency: the genotype-phenotype association. Blood Rev 2007; 21: 217-231. [ Links ]
4. Min-Oo G, Gros P. Erythrocyte variants and the nature of their malaria protective effect. Cell Microbiol 2005; 7: 753-763. [ Links ]
5. Beutler E. Red Cell Metabolism. A Manual of Biochemical Methods. 2nd ed. New York: Grune & Stratton, 1975. [ Links ]
6. Talmud P, Tybjaerg-Hansen A, Bhatnagar D, et al. Rapid screening for specific mutations in patients with a clinical diagnosis of familial hypercholesterolaemia. Atherosclerosis 1991; 89: 137-141. [ Links ]
 Correspondence:
Correspondence:
P Durand
(pierre.durand@wits.ac.za)
Accepted 7 November 2007.

