










Serviços Personalizados
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkSAMJ: South African Medical Journal
versão On-line ISSN 2078-5135
versão impressa ISSN 0256-9574
SAMJ, S. Afr. med. j. vol.98 no.3 Pretoria Mar. 2008
ORIGINAL ARTICLES
Huntington's disease: Genetic heterogeneity in black African patients
D S MagaziI; V BonevII; M MoagiBIII; Z IqbalIV; M DludlaV; C H van der MeydenVI; A KrauseVII
IMB BCh, MMed (Neurol), FCP (Neurol) (SA). Department of Neurology, Dr George Mukhari Hospital, University of Limpopo (Medunsa campus), Gauteng
IIMB ChB, FCN (SA). Department of Neurology, Dr George Mukhari Hospital, University of Limpopo (Medunsa campus), Gauteng
IIIMB Ch. Department of Neurology, Dr George Mukhari Hospital, University of Limpopo (Medunsa campus), Gauteng
IVBSc, MBBS. Department of Neurology, Dr George Mukhari Hospital, University of Limpopo (Medunsa campus), Gauteng
VMB ChB. Department of Neurology, Dr George Mukhari Hospital, University of Limpopo (Medunsa campus), Gauteng
VIMB BCh, FCP (SA), MD. Department of Neurology, Dr George Mukhari Hospital, University of Limpopo (Medunsa campus), Gauteng
VIIMB BCh, PhD. Division of Human Genetics, School of Pathology, National Health Laboratory Service and University of the Witwatersrand, Johannesburg
ABSTRACT
OBJECTIVE: Huntington's disease (HD) has been reported to occur rarely in black patients. A new genetic variant-Huntington's disease-like 2 (HDL2) - occurring more frequently in blacks, has recently been described. The absence of an expanded trinucleotide repeat at the chromosome 4 HD locus was previously regarded as a way of excluding classic HD. The objective of this paper is to describe a number of black patients with genetically proven HD and to review its occurrence in Africa.
METHODS: Eleven black families (12 subjects), with genetically proven HD, are described: 9 from the Dr George Mukhari Hospital, and 2 from private practice in Tshwane.
RESULTS: Chorea was present in all 12 patients and cognitive decline in 9. Nine had an age of onset between 30 and 50 years. Six families exhibited expansion of the trinucleotide repeat at the chromosome 4, IT 15 gene (HD), and 5 a junctophilin (JPH3) trinucleotide expansion at chromosome 16 (HDL2). The HDL2 subtype showed a tendency towards a later age of onset.
CONCLUSIONS: The clinical presentation of the two genotypes (i.e. HD and HDL2) appears to be similar. The actual rate of occurrence of HD in blacks may require re-assessment. Considering the number of Huntington's chorea patients occurring in our area (Garankuwa), the possibility of clustering of the condition arises.
George Huntington's description of Huntington's disease (HD) in 1872 (at the age of 22 years) remains the basic pillar of diagnosis: 'A hereditary chorea, tendency to insanity and suicide and its manifesting itself as a grave disease in adulthood'.1 HD is a progressive autosomal dominant disorder, characterised by involuntary choreiform movements, psychiatric manifestations with cognitive decline and, rarely, a bradykinetic rigidity. A G8 HD probe developed in Boston for preclinical and prenatal screening of HD using molecular genetics was first acquired in South Africa in the late 1980s.2 The disease has been shown to be due to an increased number of trinucleotide repeats in the IT 15 gene on chromosome 4p 16.3. Measurement of the CAG trinucleotide expansion has been found to be a highly sensitive and specific marker for the diagnosis of HD (sensitivity 98.8%; 95% confidence interval (CI) 97.7 - 99.4%; specificity 100%, 95% CI 95.2 - 100%).3 In view of the excellent sensitivity and specificity, it has been suggested that the absence of CAG repeats at chromosome 4 excludes the diagnosis of HD.4
Harper, in an article on the epidemiology of HD, suggested the presence of a separate mutation in people of black African origin.5 HD is considered rare among South African blacks, with an estimated prevalence rate of 1 per 10 million.6 However, this assessment was before genetic tests became available for the condition. Workers from Johns Hopkins Hospital in Baltimore, USA, have since demonstrated a repeat expansion mutation of CAG/CTG in the junctophilin-3 gene on chromosome 16q 23-24 associated with Huntington's diseaselike 2 (HDL2) in 1 Moroccan and 4 African-American subjects.7 The present paper reports on the clinical and genetic features in 11 black African families and briefly reviews the literature on HD in blacks in Africa.
Methods
Eleven families (12 subjects) with genetically diagnosed HD were identified and described: 9 were seen at Dr George Mukhari Hospital (between January 2004 and December 2005) and 2 were from private practice in Tshwane, with 1 of the latter families seen between 2001 and 2002. The genetic tests were performed at the Division of Human Genetics, School of Pathology, National Health Laboratory Service (NHLS) and University of the Witwatersrand, Johannesburg. Every patient was assessed clinically by a qualified neurologist. A Mayo Mini Mental test8 was performed on 9 subjects. Some of the investigations included brain imaging, copper studies, thyroid functions, syphilis, HIV and other tests in selected patients. We also undertook a review of literature on the occurrence of HD in Africa.
Results
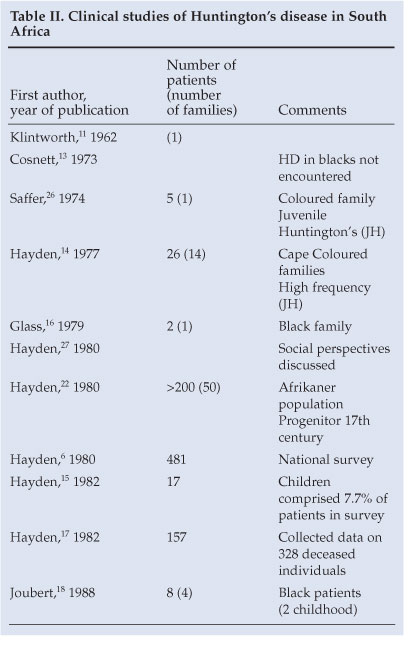
The clinical and genetic data are summarised in Table I. Six patients showed the clinical triad of an autosomal dominant family history, chorea and dementia. Nine of the 12 subjects presented with a combination of chorea and dementia. Chorea affecting all 4 limbs was present in every subject. The facial and neck movements were not observed in 1. The dementia was of a moderate to severe degree, and no patient had a picture of bradykinetic rigidity. No history of mixed ancestry could be obtained.
Six patients had an IT 15 mutation on chromosome 4 (HD1), and another 6 had a JPH3 mutation (HDL2). Those patients with a JPH3 (HDL2) abnormality tended to have a later age of onset for the chorea (average age of onset 50.5 years compared with 39 years for the IT 15 mutation). There was otherwise no recognisable difference in clinical presentation between the patients with HD1 and those with HDL2. We were unable to obtain a positive family history in 4 patients, who were proven genetically to suffer from the condition (2 with HD1, and the other 2 with HDL2).
Discussion
The diagnosis of HD has rested mainly on the clinical triad of an autosomal dominant family history, a history of progressive chorea, and dementia (usually presenting in middle adult life).1 The most consistent finding in our series was the presence of chorea which, because of its conspicuous nature, might have prompted the patients to seek medical attention. Two patients presented with normal cognitive function even though diagnosed positively on genetic testing (we expect that cognitive decline may develop with time). The ratio of the occurrence of HD and HDL2 was 1:1 among our patients. Although the numbers are small, they do suggest a higher occurrence of the HDL2 variant in this population than has been generally held. However, further studies and a larger number of black patients will be necessary before we can make definitive conclusions. There seems to be no clear difference in clinical presentation between the two genotypes except that the age of onset of chorea in the HDL2-type patients showed a tendency to be later than in the HD subjects. None of our patients presented with a bradykinetic rigidity syndrome, as may be seen in the Westphal variant; however, larger numbers of black patients are needed for further investigations.
Having undertaken a detailed enquiry, we found that 4 patients had an apparently negative family history. De novo mutations in HD have rarely been described but do not appear to be a well-recognised phenomenon.9,10 It is more likely that information was missing in the patients' histories that may require further evaluation.
The discovery of a second genetic locus, resulting in HDL2,7 suggests the need for reassessment of the actual occurrence of HD in black patients, with its associated implications for informed genetic counselling. The junctophilin-3 (JPH3) gene encodes a protein thought to be involved in the formation of junctional membrane structures and in the regulation of intracellular calcium.11
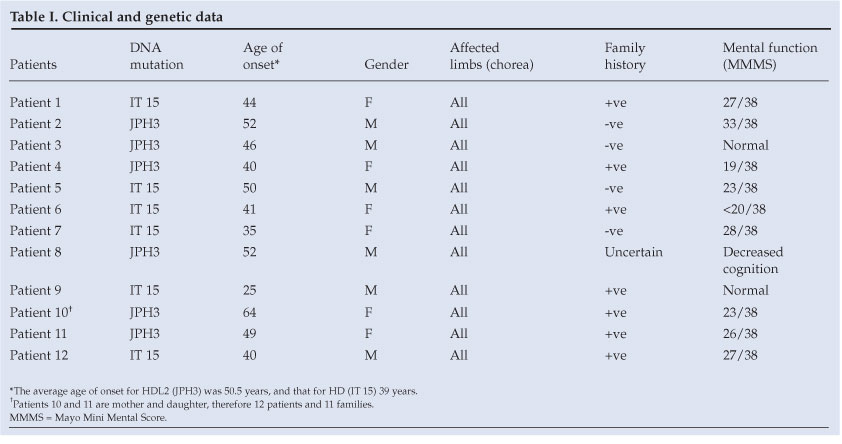
A synopsis of disease reports on clinical HD in South Africa appears in Table II. A report of a suspected patient with HD in a Zulu family appeared in 1962.12 A doyen of South African neurology commented in 1973 that he had not encountered Huntington's chorea in black patients.13 The minimum prevalence of the disease in the coloured population was reported to be 3.5/100 000;14 16% of these patients had an age of onset <20 years. The incidence of juvenile HD was found to be higher in this group than in the white population, and in fact to be one of the highest in the world.14,15 In 1979, Glass and Saffer reported Huntington's chorea in a black family.16 The comment was made that the family was probably of mixed ancestry and, quoting the authors, 'The impression gained from the literature that Huntington's chorea rarely occurs in Blacks is strengthened'. In a South African national survey reported in 1980,6 481 persons, including only 11 blacks, were identified as having died of or been afflicted with the disorder at the time. A prevalence rate of 0.1 per million was estimated in the South African 'Negro' population - much lower than the estimate of 22 per million in the white and coloured groups.6 A subsequent national survey on HD identified only 3 black patients out of a population of 19 million.17 The 4 black families with HD reported by Joubert18 emanated from a similar Medunsa geographical drainage area as our present report. This link may possibly represent a form of clustering of patients, as was suggested to have occurred in Tanzania19 and also in a survey from Maryland, USA,20 where 61 black patients with HD were recorded with a high point prevalence of 6.37/100 000. Ninety-eight black patients with HD were reported from South Carolina.21
HD was reported to be prevalent among the Afrikaner population of South Africa;22 the origin of the gene in that population has been traced over 14 generations from the present to the days of the first free burghers at the Cape of Good Hope in the late 17th century. Over 200 affected individuals in more than 50 supposedly unrelated families have been found to be ancestrally related through a common progenitor from that period.22
A synopsis of clinical HD reports from the rest of Africa is shown in Table III. Scrimgeour and Simpson,23 in a literature survey on HD in Africa, refer to reports from Kenya, Northern Tanzania, Nigeria, Togo, Zimbabwe and Ghana, and suggested that there could well be foci of the disease in Africa that were as yet unidentified for various reasons.
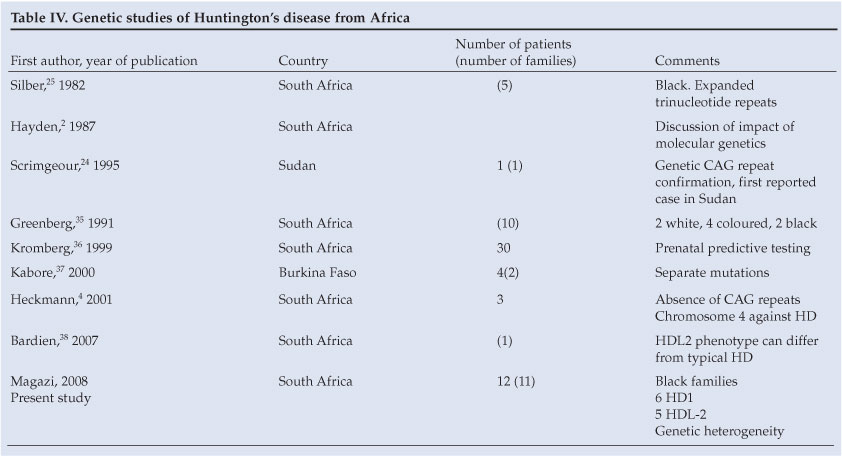
Table IV presents a summary of genetic studies on HD from Africa. Scrimgeour et al.24 and Silber et al.25 reported genetically confirmed HD patients in Africa. Heckmann et al.4 drew attention to the possible value of negative CAG trinucleotide testing on chromosome 4. Our paper supports the presence of a genetic heterogeneity involving two loci in patients with a clinical HD phenotype, making it important to assess patients clinically and to test for both HD and HDL2 before making a diagnosis. Furthermore, HD is still reported less frequently in black patients than in white patients. There is a need to reassess the prevalence estimates now that definitive genetic testing is available.
References
1. Huntington G. On chorea (an essay read before the Meigs and Mason Academy of Medicine at Middleport, Ohio, February 1872). Barbeau A, Chase NT, Paulson GW, eds. Advances in Neurology. New York: Raven Press, 1973: 33-35. [ Links ]
2. Hayden MR, Goldblatt J, Wallis G, Winship IM, Beighton P. Molecular genetics and Huntington's disease. The South African situation. S Afr Med J 1987; 71: 683-686. [ Links ]
3. Kremer B, Goldberg P, Andrew SE et al. A worldwide study of the Huntington's disease mutation: the sensitivity and specificity of measuring CAG repeats. N Engl J Med 1994; 330: 1401-1406. [ Links ]
4. Heckmann JM, Bryer A, Greenberg LJ. When is it not Huntington's disease? S Afr Med J 2001; 91(2): 132-133. [ Links ]
5. Harper PS. The epidemiology of Huntington's disease. Hum Genet 1992; 89: 365-376. [ Links ]
6. Hayden MR, MacGregor JM, Beighton PH. The prevalence of Huntington's chorea in South Africa. S Afr Med J 1980; 58: 193-196. [ Links ]
7. Holmes SE, O'Hearn E, Rosenblatt A, et al. A repeat expansion in the gene encoding junctophilin - 3 is associated with Huntington disease-like 2. Nat Genet 2001; 29: 377-378. [ Links ]
8. Kokmen E, Naessens JM, Offord KP. A test of mental status: a description and preliminary results. Mayo Clin Proc 1987; 62 (4): 281-288. [ Links ]
9. Bozza A, Malagu S, Calzolari E, Novelletto A, Pavoni M, del Senno L. Expansion of a (CAG)n repeat region in a sporadic case of HD. Acta Neurol Scand 1995: 92: 132-134. [ Links ]
10. Watanabe M, Satoh A, Kanemoto M, Ohkoshi N, Shoji S. De novo expansion of a CAG repeat in a Japanese patient with sporadic Huntington's disease. J Neurol Sci 2000; 178: 159-162. [ Links ]
11. Takeshima H, Komazaki S, Nishi M, Iino M, Kangawa K. Junctophilins: a novel family of junctional membrane complex proteins. Mol Cell 2000; 6(1): 11-22. [ Links ]
12. Klintworth GK. Huntington's chorea in South Africa. A preliminary communication drawing attention to its frequent occurrence. S Afr Med J 1962; 36: 896-898. [ Links ]
13. Cosnett JE. Neurological disease in Natal. In: Spillane JD, ed. Tropical Neurology. London: Oxford University Press, 1973: 259-272. [ Links ]
14. Hayden MR, Beighton P. Huntington's chorea in the Cape Coloured community of South Africa. S Afr Med J 1977; 52: 886-888. [ Links ]
15. Hayden MR, Macgregor JM, Saffer DS, Beighton PH. The high frequency of juvenile Huntington's chorea in South Africa. J Med Genet 1982; 19: 94-97. [ Links ]
16. Glass J, Saffer DS. Huntington's chorea in a black family. A report of 2 cases. S Afr Med J 1979; 56: 685-688. [ Links ]
17. Hayden MR, Beighton P. Genetic aspects of Huntington's chorea. Results of a national survey. Am J Med Genet 1982; 11: 135-141. [ Links ]
18. Joubert J, Botha MC. Huntington disease in South African blacks. A report of 8 cases. S Afr Med J 1988; 73: 489-494. [ Links ]
19. Scrimgeour EM. Huntington's disease in Tanzania. J Med Genet 1981; 18: 200-203. [ Links ]
20. Folstein SE, Chase GA, Wahl WE, McDonnell AN, Folstein MF. Huntington disease in Maryland: clinical aspects of racial variation. Am J Hum Genet 1987; 41: 168-179. [ Links ]
21. Wright HH, Still CN, Abramson RK. Huntington's disease in black kindreds in South Carolina. Arch Neurol 1981; 38: 412-414. [ Links ]
22. Hayden MR, Hopkins HC, Macrae M, Beighton PH. The origin of Huntington's chorea in the Afrikaner population of South Africa. S Afr Med J 1980; 58: 197-200. [ Links ]
23. Scrimgeour EM, Simpson SA. Huntington disease in black African populations (letter). Hum Genet 1992; 90(1-2): 186-187. [ Links ]
24. Scrimgeour EM, Samman Y, Brock DJ. Huntington's disease in a Sudanese family from Khartoum. Hum Genet 1995; 96(5): 624-625. [ Links ]
25. Silber E, Kromberg J, Temlett JA, Krause A, Saffer D. Huntington's disease confirmed by genetic testing in five African families. Mov Disord 1998; 13(4): 726-730. [ Links ]
26. Saffer DS, Nathan DC, Kahle PA, Steingo B. Huntington's disease in a coloured family. S Afr Med J 1974; 48: 2399-2402. [ Links ]
27. Hayden MR, Ehrlich R, Parker H, Ferera SJ. Social perspectives in Huntington's chorea. S Afr Med J 1980; 58: 201-203. [ Links ]
28. Gordon HL. Huntington's chorea in an East African. Proc R Soc Med 1935; 29: 1469-1470. [ Links ]
29. Hutton PW. Neurological disorders in Uganda. East Afr Med J 1956: 33: 209-223. [ Links ]
30. Harries JR. Neurological disorders in Kenya. In: Spillane JD, ed. Tropical Neurology. London: Oxford University Press, 1973: 207-222. [ Links ]
31. Samuels BL, Gelfand M. Huntington's chorea in a Black Rhodesian family. S Afr Med J 1978; 54: 648-651. [ Links ]
32. Scrimgeour EM. The Huntington's chorea register of Tanzania. East Afr Med J 1982; 59(4): 280-282. [ Links ]
33. Aiyesimoju BO, Osuntokun BO, Bademosi O, Adeuja AO. Hereditary neurodegenerative disorders in Nigerian Africans. Neurology 1984; 34: 361-362. [ Links ]
34. Scrimgeour EM, Pfumojena JW. Huntington disease in black Zimbabwean families living near the Mozambique border. Am J Med Genet 1992; 44: 762-766. [ Links ]
35. Greenberg LJ, Martell RW, Theilman J, Hayden MR, Joubert J. Genetic linkage between Huntington disease and the D4S10 locus in South African families: further evidence against non-allelic heterogeneity. Hum Genet 1991; 87: 701-708. [ Links ]
36. Kromberg JGR, Krause A, Spurdle AB, et al. Utilisation of predictive prenatal and diagnostic testing for Huntington's disease in Johannesburg. S Afr Med J 1999; 89(7): 774-778. [ Links ]
37. Kabore J, Ouedraogo A. Huntington disease in Burkina Faso. Rev Neurol (Paris) 2000;156(12): 1157-1158. [ Links ]
38. Bardien S, Abrahams F, Soodyall H, et al. A South African mixed ancestry family with Huntington disease like 2: Clinical and genetic features. Mov Disord 2007; 22(14): 2083-2089. [ Links ]


{kind=link}
{kind=link}
{kind=link}