










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSAMJ: South African Medical Journal
On-line version ISSN 2078-5135
Print version ISSN 0256-9574
SAMJ, S. Afr. med. j. vol.98 n.2 Pretoria Feb. 2008
GUIDELINE
Guideline for the Treatment of Haemophilia in South Africa
Johnny N MahlanguI; Anne GilhamII
IBSc (Lab Med), MB BCh, FCPath (SA), Cert Clin Haem (SA); Haemophilia Comprehensive Care Centre, Johannesburg Hospital, and Department of Molecular Medicine and Haematology, Faculty of Health Sciences, School of Pathology of the National Health Laboratory Services and University of the Witwatersrand, Johannesburg, South Africa
IIHaemophilia Comprehensive Care Centre, Johannesburg Hospital
ABSTRACT
This guideline has been prepared by the authors for and on behalf of the Medical and Scientific Advisory Council (MASAC) of the South African Haemophilia Foundation to facilitate the appropriate management of individuals with haemophilia in South Africa. Individuals with haemophilia and their physicians should be advised by a Comprehensive Haemophilia Treatment Centre. Strategies that help to prevent bleeds include regular exercise to strengthen muscles, protect joints and improve fitness; maintaining a healthy body weight to avoid extra stress on joints; and avoiding contact sports. Acute bleeds should be treated early, ideally within 2 hours of onset. Patients with mild or moderate haemophilia A may be treated with desmopressin. Bleeding in patients with severe haemophilia A without inhibitors should be treated with factor VIII concentrate. Bleeding in patients with haemophilia B without inhibitors should be treated with factor IX replacement. Tranexamic acid can be used for mucous membrane bleeding in surgical or dental procedures. Bleeds in patients with inhibitors must be managed in consultation with a haemophilia treatment centre. Major bleeding episodes are large muscle or joint bleeds, bleeds resulting from severe injury, or bleeds that affect the central nervous system; gastrointestinal system; neck or throat; hip or iliopsoas; or the forearm compartment. These bleeds may cause death or musculoskeletal deformities, and advice on their treatment should be sought from a haemophilia treatment centre physician. Appropriate factor replacement therapy must be started urgently for major bleeds, and hospitalisation is usually required to maintain adequate factor levels.
1. Introduction
This guideline has been prepared by the authors for and on behalf of the Medical and Scientific Advisory Council (MASAC) of the South African Haemophilia Foundation (see Appendix I for list of current members) to be used as a general guide to facilitate the appropriate management of individuals with haemophilia and associated complications in South Africa. The guideline recommendations are based on a number of resources, including:
- available published literature evidence on haemophilia management
- the international best practice of haemophilia care incorporating recommendations of the Association of Haemophilia Clinic Directors of Canada (AHCDC) (1999);1 the United Kingdom Haemophilia Centre Doctors' Organisation (UKHCDO) (2003);2 the World Federation of Haemophilia (WFH) (2005);3 the Medical Advisory Committee of the Haemophilia Foundation of New Zealand (2005);4 the Medical and Scientific Advisory Council of the National Haemophilia Foundation of the USA (2006);5 and the Australian Health Ministers' Advisory Council (AHMAC) (2006)6
- and, where published empirical evidence is unavailable, the consensus expert opinion of the Medical and Scientific Advisory Council members and the international haemophilia fraternity, taking into account the availability of expertise and treatment products in South Africa.
This guideline has been designed to provide practical and accessible guidance for primary care health care practitioners, who might not be very familiar with haemophilia management, as well as to summarise best practice for practitioners at the secondary and tertiary health care levels. The ultimate responsibility for the care and choice of treatment of patients with haemophilia lies with the attending doctor and the patient being treated. Therefore, the guideline is not a substitute for the attending doctor's clinical judgement. It should not be considered to encompass management of every patient in every clinical situation.
This guideline does not attempt to cover the cost-effectiveness of different treatments for haemophilia. Although it is a rare disease, haemophilia care is expensive owing to the high costs of regular factor replacement to treat bleeds. It is therefore important that resources be used optimally and in the safest and most effective manner. World Federation of Haemophilia data show that treatment of persons with haemophilia outside haemophilia treatment centres results in higher mortality and cost compared with treatment in the haemophilia treatment centres. The care of people with haemophilia often requires a multidisciplinary team to address different aspects of patient problems. The World Health Organization and the World Federation of Haemophilia recommend that this disease be managed in association with a haemophilia comprehensive care centre (see Appendix II for a list of haemophilia treatment centres in South Africa). Furthermore, all aspects of haemophilia disease management, including rheumatology, orthopaedic surgery, dentistry, clinical genetics, infectious diseases, physiotherapy and gynaecology, should be managed in consultation with a haemophilia specialist.
2. Haemophilia overview
Haemophilia refers to inherited bleeding disorders caused by deficiency of specific coagulation factors. Haemophilia A is caused by coagulation factor VIII (FVIII) deficiency, haemophilia B by deficiency of coagulation factor IX (FIX), and haemophilia C by deficiency of coagulation factor XI. These clotting factor deficiencies are caused by recessive mutations of the respective clotting factor genes. As the recessive mutant FVIII and FIX genes are located on the X chromosome, both haemophilia A and haemophilia B are inherited in an X-linked pattern. Consequently, in these conditions, males are affected and females are carriers of haemophilia. Both diseases have the same clinical presentation, so their specific diagnosis must be established by factor assay. Haemophilia A has a prevalence of about 1 in 10 000 males, while haemophilia B is less common, with a prevalence of about 1 in 35 000 males.7 The combined prevalence of both haemophilia A and B has been estimated as approximately 1 in 5 000 live male births.7 Haemophilia C is an autosomally inherited condition with a high prevalence in Ashkenazi Jews and is uncommon in the general population of South Africa, and will therefore not be discussed further in this guideline.
The FVIII gene spans 186 kb, contains 26 exons, and is located on the long arm of the X chromosome at Xq28.8 This gene is unusual because it contains two additional genes, F8A and F8B, within intron 22. The most common mutation in haemophilia A is a large inversion and translocation of exons 1 - 22 (together with introns) away from exons 23 - 26, the mechanism of which is homologous recombination between the F8A gene in intron 22 and one of the F8A copies located outside the FVIII gene.9,10 This intron 22 inversion results in no FVIII protein being produced and is responsible for about 40% of cases of severe haemophilia A.11 This inversion arises almost exclusively in male germ cells.12 A similar inversion involves intron 1 of the FVIII gene, and has a prevalence of approximately 5% in patients with severe haemophilia A.13 Other mutations causing haemophilia A are mainly single-base substitutions, with over 600 such mutations having been described. Smaller numbers of sequence deletions and insertions have also been reported. A database of haemophilia A mutations is available at http://europium.csc.mrc.ac.uk/.
The FIX gene spans 33.5 kb, contains 8 exons, and is located on the long arm of the X chromosome at Xq27.14 Haemophilia B results from a wide range of heterogeneous mutations spread throughout the FIX gene.15 Most of these mutations are single-base substitutions. Studies in various populations have found evidence of a founder effect, where large numbers of cases of mild haemophilia B are due to a small number of founder mutations.16-19 A database of haemophilia B mutations is available at http://www.kcl.ac.uk/ip/petergreen/haemBdatabase.html. About 30% of individuals with haemophilia (either A or B) represent spontaneous mutations and have a negative family history.20
3. Clinical manifestation of haemophilia
The clinical manifestation of bleeding in haemophilia depends on the severity of the disease. Disease severity is classified as severe, moderate and mild depending on the level of coagulation factor in blood as shown in Table I.21 The assessment of bleeding requires a systematic approach including a detailed medical history and physical examination.
Haemophilia should be suspected in individuals presenting with:
- A family history of bleeding, particularly in males
- A lifelong history of easy bruising
- Spontaneous bleeding into joints, subcutaneous soft tissues and mucous membranes
- Excessive bleeding following haemostatic challenge (e.g. trauma, surgery, etc.).
The bleeding history should be detailed and should include enquiry relating to:
- Age of the patient when bleeding started
- Site of bleeding (joints, skin, mucous membranes, etc.)
- Type of bleeding (petechiae, purpura, haematoma, etc.)
- Extent of bleeding (localised, systemic)
- Induced or spontaneous bleeding
- Immediate or delayed bleeding.
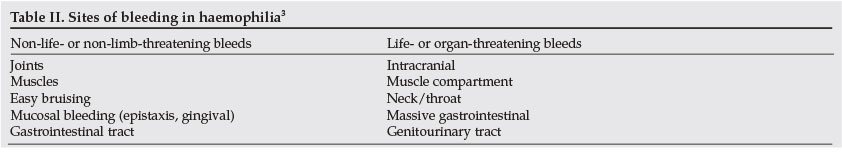
While haemophilia is a systemic haemorrhagic disorder, haemophilic bleeds occur in fairly limited sites, as shown in Table II. Life-threatening and potentially organ-threatening bleeds are the most challenging. It is fortunate that the latter bleeds are also uncommon, as illustrated in Table III. The most frequent bleeds in haemophilia are haemarthrosis, followed by muscle and subcutaneous haematomas. As summarised in Table IV, knee haemarthroses are the commonest joint bleeds, followed by the elbows, ankles, shoulders, wrists and hips, in that order. Why joints are targets for bleeding in individuals with haemophilia remains a mystery. Some of the postulated but unproven hypotheses include high fragility of synovial membrane blood vessels, increased fibrinolysis and decreased thromboplastin in the synovial fluid.22 There is poor correlation between normal joint physical activity and frequency of bleeding.


The aim of physical examination in haemophilia is to:
- Establish the site of bleeding
- Establish the extent of bleeding and whether bleeding is life-, limb- or organ-threatening
- Exclude non-coagulopathic bleeding, i.e. bleeding due to platelet and blood vessel abnormalities.
The physical examination hallmarks of an acute haemophilic bleed are swelling, pain and limited range of joint motion. Chronicity of bleeding diathesis is indicated by muscle atrophy, joint axial deformity, crepitations, joint instability and flexion contractures.
4. Diagnosis and classification
It is important to make an accurate diagnosis of haemophilia as this will dictate the nature of therapeutic intervention. The essential elements for making a diagnosis include:
- A comprehensive bleeding history (see above)
- A complete physical examination (see above)
- Performing screening tests for bleeding diathesis
- Performing confirmatory tests.
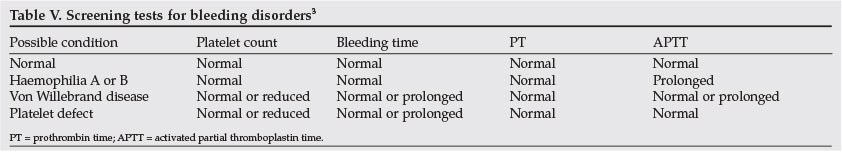
For any patient presenting with a bleeding diathesis, the screening tests to be performed will be dictated by the clinical findings. These screening tests include measurement of platelet count, international normalised ratio (INR) and activated partial thromboplastin time (aPTT). Bleeding time should be determined if bleeding is suspected to be due to platelet dysfunction or blood vessel abnormality. Table V illustrates the diagnostic interpretation of the screening tests.
The screening tests are followed by confirmatory tests, which include specific factor assays, inhibitor assays, platelet function tests and von Willebrand factor assays, where indicated. These tests are available at the haemophilia comprehensive care centres. Since haemophilia A and B are indistinguishable clinically and on screening tests, their diagnosis must be confirmed by specific factor assays.
Factor assays allow classification of haemophilia as severe, moderate or mild depending on the plasma concentration of FVIII or FIX (Table I).21 The clinical severity of haemophilia is inversely related to circulating clotting factor.
5. General principles of management of people with haemophilia
- Manage people with haemophilia at or under the supervision of a haemophilia comprehensive care centre (level III-3 evidence (see Table VI for designation of levels of evidence)).23 As far as necessary, all members of the multidisciplinary haemophilia care team should be involved.
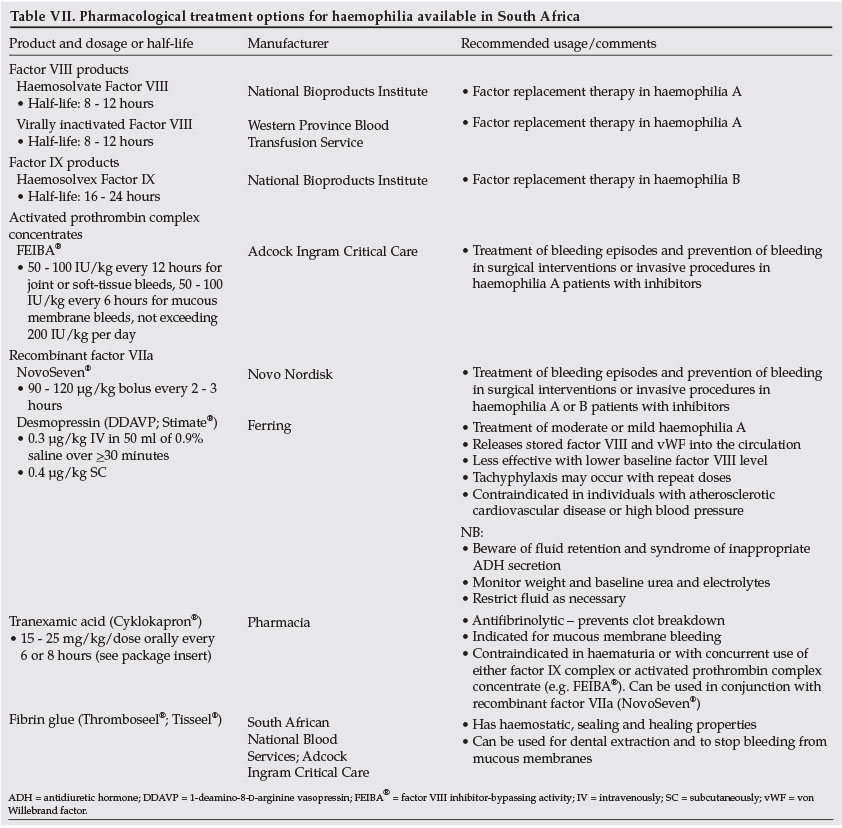
- Treat specific deficiencies with specific deficient concentrate (see Table VII for details of factor products, as well as other pharmacological treatments for haemophilia, available in South Africa). Avoid fresh-frozen plasma if specific factor concentrate is available (level III-3 evidence).24,25
- Treat bleeds early (level III-3 evidence).26,27 All patients should be taught factor self-administration at home under supervision and monitoring by a haemophilia specialist.
- Treat bleeds first, if diagnosis is known, before sending patients for further investigative procedures (level IV evidence).
- Avoid antiplatelet drugs, in particular aspirin and non-steroidal anti-inflammatory drugs (level IV evidence).
- Avoid all intramuscular injections (level III-3 evidence).28,29
- Give adequate pain relief, especially for large joints and muscle bleeds. Use paracetamol, cyclo-oxygenase-2 (COX-2) inhibitors or opioid analgesics (level III-3 evidence).30
- Institute adjunct therapeutic measures as soon as possible (level IV evidence). These include:
- rest of the affected limb in a functional position
- ice application
- immobilisation
- compression bandage if applicable
- elevation of the affected limb.
Vaccinate all individuals with haemophilia with appropriate vaccines (level III-3 evidence).31,32
- Standard public vaccination for children can be given without factor concentrate prophylaxis. Application of ice and prolonged pressure for 10 minutes is recommended. Prophylactic cover for irritant vaccines such as tetanus should be given.
- Travel vaccination does not require prophylaxis.
- All individuals with haemophilia who are seronegative for hepatitis B should be vaccinated and seroconversion monitored.
- All individuals with haemophilia who are hepatitis A IgG-negative should be vaccinated with hepatitis A vaccine.
Institute primary or secondary prophylaxis where appropriate (level 1 evidence).33
- Primary prophylaxis is factor infusions given to prevent bleeding and its consequences, usually starting in the first or second year of life, before the third bleed.33
- Primary prophylaxis should be considered for infants with severe haemophilia who are at high risk of developing haemophilic arthropathy.
- Secondary prophylaxis is factor infusions given to prevent recurrent joint bleeds after target joint bleeding. Secondary prophylaxis can be given intermittently prior to activities likely to cause bleeding, or
- Secondary prophylaxis should be used to manage chronic synovitis prior to synovectomy.
- Single-dose secondary prophylaxis should be given prior to an event likely to result in bleeding.
- Continuous secondary prophylaxis without a definite end point is not recommended (level IV evidence).
- Encourage healthy lifestyles and other measures to prevent bleeding (level III-3 evidence).34-36
- Recommend regular exercise to people with haemophilia to strengthen the muscles around joints.
- Encourage maintenance of healthy body weight.
- Discourage participation in contact sports such as rugby and football.
- Look for and actively manage infectious and immunological complications of haemophilia treatment (level IV). Screen for inhibitors regularly, at least twice a year, particularly when factor replacement has reduced efficacy.
Manage individuals who have haemophilia with inhibitors with appropriate bypassing agents or tolerisation.
Screen for exposure to HIV and hepatitis at least twice a year in patients exposed to plasma-derived products not virucidally treated.
Treat transfusion-transmitted and other viral infections in haemophilia.
6. Treatment of haemophilia A without inhibitors
6.1 Treatment aims
- To achieve a plasma FVIII level of 80 - 100% for major bleeds and 40 - 60% for minor bleeds.
- To continue treatment until bleeding stops.
6.2 Treatment products (refer to Table VII for further details)
- Desmopressin (DDAVP; 1-deamino-8-D-arginine vasopressin (Stimate®)) for mild/moderate haemophilia A.
- pdFVIII - plasma-derived, solvent detergent-treated (Haemosolvate Factor VIII concentrate or WPBTS AHF FVIII concentrate).
- Recombinant human FVIII (rhFVIII) - in the process of registration with the regulatory authority in South Africa.
- Antifibrinolytic agent (tranexamic acid (Cyklokapron®)).
- Adjunctive agents (fibrin glue (Thromboseel®; Tisseel®)).
6.3 Treatment regimen
- DDAVP for patients with mild or moderate haemophilia A shown to be DDAVP responders:
- 0.3 µg/kg IV or subcutaneous.
- Intranasal DDAVP dose is 300 ug for adults and 150 ug for children.
- DDAVP may be administered once every 24 hours. If given for more than 3 consecutive days, repeated doses may lead to tachyphylaxis.
- pdFVIII concentrate:
- Minor bleeds should be treated with 20 - 40 IU/kg IV 12-hourly.
- Major bleeds should be treated with 40 - 50 IU/kg 12-hourly.
- If symptoms persist after 24 hours, check for inhibitors. If inhibitors are absent, continue with this regimen for 24 hours until the symptoms settle. If inhibitors are present, treat as per inhibitor patient with bypassing agents.
- Recombinant FVIII concentrate:
- Given IV rFVIII in the same doses as pdFVIII.
- Tranexamic acid:
- For mucous membrane-based bleeding given as 25 mg/kg every 8 hours by mouth.
- Contraindicated in patients with urinary tract bleeds and concurrent use of aPCC (FEIBA®).
- Fibrin glue (Thromboseel®):
- Topical agent for accelerating haemostasis and wound healing.
- Mixture of platelet-rich plasma and thrombin.
- Applied on wound at 0.2 ml/cm2.
6.4 Treatment notes for haemophilia without inhibitors
- Both pdFVIII and rhFVIII are efficacious in the treatment of bleeding episodes in haemophilia A (level II evidence).37
- There has been no new non-enveloped viral infection transmission (HIV, hepatitis B or hepatitis C) reported with the use of intermediate purity pdFVIII concentrate used in South Africa (level IV evidence). A case of hepatitis A infection has been reported.
- 1 IU/kg of pdFVIII or rhFVIII will increase in vivo FVIII level by 2% (level II evidence).37
- The rate of inhibitor induction by pdFVIII and rhFVIII appears to be equivalent (level III-3 evidence).38
- The therapeutic efficacy of intravenous and subcutaneous DDAVP is equivalent (level II evidence).39
- Giving DDAVP for more than 3 consecutive days may lead to reduction in responsiveness (tachyphylaxis) (level IV evidence).40
- Test doses of DDAVP and FVIII should be performed to demonstrate efficacy (level IV evidence).41
- Fibrin glue (Thromboseel®) is a plasma-derived product the efficacy of which has not yet been established in clinical trials.
7. Treatment of haemophilia B without inhibitors
7.1 Treatment aim
- To achieve a plasma FIX level of 60 - 80% for major bleeds and 20 - 40% for minor bleeds.
- Treatment should continue until bleeding stops.
7.2 Treatment products
- Plasma-derived FIX (pdFIX; Haemosolvex Factor IX)
- Antifibrinolytics (tranexamic acid).
7.3 Treatment regimen
- Plasma-derived, intermediate purity FIX:
- Minor bleeds should be treated with 20 - 40 IU/kg IV once or repeated at 24-hour intervals.
- Major bleeds should be treated with 60 - 80 IU/kg IV daily.
- Tranexamic acid:
- For mucous membrane-based bleeding given as 15 - 25 mg/kg every 8 hours by mouth.
- Contraindicated in patients with urinary tract bleeds.
- Do not use concurrently with an aPCC (FEIBA®).
- Fibrin glue (Thromboseel®):
- Topical agent for accelerating haemostasis and wound healing.
- Mixture of platelet-rich plasma and thrombin.
- Applied on wound at 0.2 ml/cm2.
7.4 Treatment notes
- pdFIX is safe and effective for the treatment of bleeding diathesis due to FIX deficiency without inhibitors (level IV evidence).
- One IU/ml of pdFIX infused will raise the plasma FIX by 1% (level II evidence).42 The half-life of FIX is 16 - 24 hours and it is therefore given once per 24 hours.
- The pdFIX is a prothrombin complex concentrate (PCC) containing other vitamin K-dependent factors (II, VII and X). Large doses of PCCs may be associated with thrombosis or disseminated intravascular coagulation (DIC) (level IV evidence).43 Haemosolvex can be used for treating warfarin overdose and factor X deficiency (level III-3 evidence).44-47
- The incidence of inhibitor induction by aPCC is extremely low (level II evidence).42
- DDAVP is ineffective for the treatment of FIX deficiency (level IV evidence).
- The efficacy of fibrin glue has not been validated in clinical settings (level IV evidence).
8. Management of patients with inhibitors
8.1 Overview of inhibitors
Inhibitors are usually IgG4 antibodies that neutralise the procoagulant activity of FVIII or FIX. About 10 - 15% of haemophilia A patients and 1 - 3% of haemophilia B patients may develop persistent inhibitors,3 which make treatment with factor concentrates difficult. Bleeds in patients with inhibitors must be managed in consultation with a haemophilia treatment centre.
Risk factors for the development of inhibitors include severe haemophilia and a family history of inhibitor development. Inhibitors are more common in black patients. If a child with haemophilia A is going to develop an inhibitor, this usually happens within the first 50 exposure days after starting FVIII replacement therapy.
Inhibitor titres are measured in Bethesda units (BU), with low-titre inhibitors measuring <5 BU and high-titre inhibitors measuring >5 BU. Inhibitor patients are further classified as high or low responders based on the way inhibitor titres change in response to treatment. In low-titre inhibitor, low responders, the inhibitor titre does not rise above 5 BU even after factor replacement therapy, and inhibitors may be transient despite continual specific factor replacement. In low-titre inhibitor, high responders, inhibitor titres rise above 5 BU after factor replacement therapy. In high-titre inhibitor, high responders, inhibitor titres are greater than 5 BU and rise further after factor replacement therapy; if not treated for a long period the level may fall, but there will be a recurrent, rapid anamnestic response 3 - 5 days after factor infusion.
All patients should be monitored every 3 - 6 months for the development of inhibitors. This is particularly important and should be done more frequently in newly diagnosed black children with severe haemophilia A, who are at greater risk. A surgical procedure in a person with haemophilia should never be undertaken without prior checking for inhibitors. Inhibitors should also be tested for if there is suboptimal response to factor replacement therapy.
If a BU test to measure the amount of inhibitors cannot be performed, the presence of an inhibitor can be checked using an APTT.48 Take a mixture of patient and normal plasma incubated together for 1 - 2 hours at 37°: in the presence of an inhibitor the APTT will be prolonged.
Immune tolerisation should be initiated at a haemophilia treatment centre. Successful therapy (eliminating the inhibitor) may take months. Several regimens are effective;49 the Dutch regimen (25 IU FVIII/kg 3 times per week) is the most affordable.50
8.2 Treatment of haemophilia A with inhibitors
8.2.1 Treatment aim
- To stop bleeding in a patient with FVIII deficiency with inhibitors.
8.2.2 Treatment products
- Recombinant FVIIa (rFVIIa; NovoSeven®)
- Activated prothrombin complex concentrate (aPCC; FEIBA®)
- High doses of pdFVIII (Haemosolvate) or WPBTS AHF
- Antifibrinolytic (tranexamic acid)
- Adjunctive agent (fibrin glue).
8.2.3 Treatment regimen for bleeding episode
- Low-responder inhibitors (<5 BU):
- Give pdFVIII at a dose of 2 - 3 times the normal dose.
- Must monitor clinical response. If there is no response and inhibitor levels have increased, treat with one of the bypassing agents.
- High-responder inhibitors:
- aPCC (FEIBA®):
- Give dose of 50 - 100 IU/kg IV 12-hourly for 3 days.
- Do not exceed a maximum dose of 200 IU/kg and do not give concurrently with antifibrinolytic drugs, because of increased risk of thrombosis.
- rFVIIa (NovoSeven®):
- Give dose of 90 - 120 ug/kg IV every 2 - 3 hours as bolus or 20 IU/kg/hour as continuous infusion.
- Antifibrinolytic can be given concurrently with rFVIIa.
8.3 Treatment regimen for inhibitor eradication
8.3.1 Treatment aims
- To remove inhibitors to undetectable level with normal FVIII recovery and half-life.
8.3.2 Treatment protocols
- Bonn protocol (modified):51
- This protocol may be suitable for young patients with a low-inhibitor titre and a short time between inhibitor development and their treatment (level IV evidence).52
- Give 150 IU/kg FVIII or 50 - 100 IU/kg FEIBA® every 12 hours until the inhibitor level drops to less than 1 BU.
- Then reduce FVIII to 150 IU/kg daily until the inhibitor is no longer detectable.
- Van Creveld protocol:50
- Give neutralising dose of 25 - 50 IU FVIII 12-hourly for 1 - 2 weeks.
- Give tolerising dose of 25 IU 3 times a week until inhibitor disappears.
- Malmö protocol:53
- This protocol may be suitable for inhibitor levels of 10 BU or more.
- Use extracorporeal immunoadsorption to reduce inhibitors to less than 10 BU.
- Give FVIII at 200 IU/kg aiming for recovery of 40 -100 IU/dl and maintained at 30 - 80 IU/dl.
- Give cyclophosphamide at 12 - 15 mg/kg IV for days 1 and 2 followed by 2 - 3 mg/kg for 10 days.
- Give intravenous immunoglobulin (IVIG) at a dose of 2.5 -5 g/kg on day 1 followed by 0.4 mg/kg on days 4 and 5.
8.3.3 Notes on inhibitor treatment regimens
- There are no available studies with data to show effectiveness of high doses of FVIII (level IV evidence). High-responder inhibitor should not be treated with high doses of FVIII (level IV evidence).54
- There is no difference in outcomes between patients tolerised with plasma-derived or recombinant FVIII (level IV evidence).55
- rFVIIa may be given as a bolus or continuous infusion (level IV evidence).56
- Both aPCC and rFVIIa appear to have equivalent efficacy (level II evidence) (FENOC study).57
- Plasmapheresis may be used to reduce high-titre inhibitors prior to infusion of FVIII (level IV evidence).58
- Immunosuppressive therapy using cyclophosphamide to reduce inhibitor levels is not recommended for treating acute bleeding episodes (level IV evidence).
8.4 Treatment of haemophilia B with inhibitors
Both rFVIIa and aPCC are effective for treatment of acute bleeding episodes in patients with high titre and/or high responder to FIX (level II evidence) (FENOC study).57 An aPCC should be carefully monitored for anaphylaxis and anamnestic reaction. Therefore patients with haemophilia B and inhibitors are best treated with rFVIIa, the only bypassing agent that does not contain FIX.59
There is no evidence to guide tolerisation procedures in patients with haemophilia B and inhibitors. Plasma-derived FIX may be used for tolerisation with careful monitoring of anaphylactic reactions.
9. Treatment of specific haemorrhages
9.1 Major and minor bleeding episodes
Major bleeding episodes are advanced muscle or joint bleeds, bleeds resulting from severe injury, or bleeds that affect the central nervous system; gastrointestinal system; neck or throat; hip or iliopsoas; or forearm compartment. These bleeds may cause death or crippling deformities, and advice on their treatment should be sought from a haemophilia treatment centre physician. Appropriate factor replacement therapy must be started urgently, and hospitalisation is usually required to maintain adequate factor levels. If the patient with a major bleeding episode has an inhibitor, the haemophilia treatment centre must be consulted.
Bleeds into the muscle or soft tissue, or mouth or gums, are considered minor bleeding episodes, as are epistaxis, painless haematuria and early joint bleeds. These bleeds should be treated early to avoid long-term complications. If there are uncertainties about medical management, a haemophilia treatment centre should be consulted.
9.2 Haemarthroses
9.2.1 Commonly affected joints
- See Table IV.
9.2.2 Clinical features
- Tingling sensation or aura - early symptom
- Joint stiffness - early symptom
- Joint pain - early/late symptom
- Limited range of motion - late sign
- Swelling - late sign.
9.2.3 Treatment goals
- In the case of haemophilia A, the aim is to achieve a peak blood FVIII level of 40 - 60% for minor bleeds and 80 - 100% for major bleeds. In the case of haemophilia B the target is 20 - 40% for minor bleeds and 60 - 80% for major bleeds.
- To rehabilitate the joint to a prebleed functional state.
9.2.4 Treatment approach
- Haemophilia A: 20 - 40 IU/kg FVIII if minor bleed or 40 -50 IU/kg IV if major bleed given twice a day.
- Haemophilia B: 20 - 40 IU/kg FIX if minor bleed or 60 -80 IU/kg IV if major bleed IV daily.
- If symptoms continue after 24 hours and there are no inhibitors, continue with treatment until symptoms settle.
- Rest affected joint/limb and immobilise with non-circumferential cast.
- Apply ice, 5 minutes on and 10 minutes off. Start ice application immediately.
- Temporarily immobilise limb with splints, slings or crutches.
- Give analgesia (paracetamol, or propoxyphene, buprenorphine or tramadol) for pain not settling after factor infusion if bleed is major.
- Start rehabilitative exercises under factor cover as soon as symptoms disappear to facilitate return to prebleed structure and function.
9.3 Muscle and soft-tissue bleeding
9.3.1 Commonly affected muscles - predilection for large flexor muscles
- Iliopsoas
- Forearm muscles
- Gastrocnemius
- Quadratus femoris muscles
- Others following intramuscular injections.
9.3.2 Clinical features
- Muscle tightness
- Painful due to compartment bleed
- Paraesthesia
- Swelling and hard to touch, and asymmetry
- Warmth and bruising
- Distal pallor and pulselessness
- Muscle dysfunction or limited range of function.
9.3.3 Treatment goals
- To raise plasma factor level to 80 - 100% if iliopsoas muscle or 60 - 80% if non-iliopsoas muscle.
- To keep peak factor level 50% until symptoms start resolving and normal function is recovering.
- Prevent further bleeds during muscle rehabilitation.
9.3.4 Treatment approach
- Admit to hospital for bed rest and close monitoring and consult a haemophilia treatment centre or haemophilia specialist.
- Haemophilia A: 40 - 50 IU/kg FVIII IV bolus 12-hourly if iliopsoas and 20 - 40 IU/kg if other muscles.
- Haemophilia B: 60 - 80 IU/kg FIX IV daily if iliopsoas and 20 - 40 IU/kg if other muscles.
- Continue with treatment until symptoms subside.
- May need surgical decompression if there is serious neurovascular compromise.
- Ultrasound or computed tomography (CT) indicated to confirm diagnosis and get baseline clot size.
- Rest affected joint/limb and immobilise with non-circumferential cast.
- Apply ice, 5 minutes on and 10 minutes off. Start ice application immediately.
- Temporarily immobilise limb with splints, slings or crutches.
- Give analgesia (paracetamol, COX-2 inhibitor, dextropropoxyphene, or opioids) for pain not settling after factor infusion if bleed is major.
- Start rehabilitative exercise under factor cover as soon as symptoms disappear to facilitate return to prebleed structure and function.
- Rehabilitation after a bleed is essential to maintain strength and range of motion. Rehabilitation exercises should be started as soon as the pain is gone, starting with static exercise. Free active exercises where the only resistance is gravity may be started 3 days after the resolution of the bleed. Weight-bearing exercises to increase muscle strength and bulk may be started 10 days after the resolution of the bleed.
9.4 Head injury and central nervous system bleeds 9.4.1 General comments
- All CNS bleeds and head injuries should be treated as medical emergencies with immediate hospitalisation of the patient.
- Patient should receive treatment with factor concentrate before further investigations are undertaken.
- CNS bleeds can be life-threatening or result in permanent nerve damage.
9.4.2 Clinical features
- Headaches, particularly those which are persistent despite analgesia
- Signs of raised intracranial pressure (nausea, vomiting, neck stiffness, photophobia)
- Blindness, deafness, dizziness, loss of balance, ataxia
- Lethargy, drowsiness, vertigo, seizures
- Loss of consciousness, confusion, irritability
- Focal neurological deficit, muscle weakness, paralysis.
9.4.3 Treatment goals
- Raise factor level to 80 - 100% if haemophilia A and 60 - 80% if haemophilia B for 7 days.
- Maintain plasma factor level at 50% if haemophilia A and 30% if haemophilia B for a further 14 days.
9.4.4 Treatment approach
- Admit to hospital.
- Must give factor replacement early to limit bleeding extent.
- Haemophilia A: give 40 - 50 IU/kg FVIII IV 12-hourly for 7 days.
- Haemophilia B: give 60 - 80 IU/kg FIX IV daily for 7 days.
- Involve neurosurgical and haematological expertise early.
- Perform urgent CT or MRI scan after treatment.
- Anti-epileptic medication as soon as bleed is confirmed.
- Monitor factor level pre- and post-infusion.
- Continuous infusion instead of bolus injection of factor concentrate may be used.
9.5 Oral bleeding
9.5.1 Common sites of bleeding
- Gingival mucosa, buccal mucosa
- Dental caries
- Bitten tongue
- Torn upper lip.
9.5.2 Clinical features
- Vomiting blood
- Small cut or laceration in mouth bleeding profusely
- Gingivitis
- Oral infection.
9.5.3 Treatment goal
- Raise factor plasma level to 20 - 40% with FVIII or FIX concentrate.
- Use adjunctive therapy in addition to factor to stop bleeding.
9.5.4 Treatment approach
- Haemophilia A: give 20 - 40 IU/kg FVIII 12-hourly IV.
- Haemophilia B: give 20 - 40 IU/kg FIX daily IV.
- Tranexamic acid solution: give 5 - 10 ml (500 mg/5 ml) 6-hourly, holding in mouth for 2 minutes before swallowing. Tranexamic acid tablets can also be crushed in warm water before swallowing.
- Continue factor infusion and tranexamic acid until bleeding stops.
- May need local measures to stop bleeding.
- Check haemoglobin level if bleeding is excessive and patient symptomatic.
9.6 Gastrointestinal bleeding
9.6.1 Sites of bleeding
- Can be anywhere along the gastrointestinal tract
- Angiodysplasia
- Bleeding peptic and duodenal ulcers
- Bleeding haemorrhoids.
9.6.2 Clinical features
- Melaena stools
- Haematochezia
- Haematemesis
- Frank fresh blood per rectum
- Abdominal pain or acute abdomen
- Abdominal swelling or obstruction.
9.6.3 Treatment goals
- Raise plasma factor level to 80 - 100% for up to 6 days.
- Maintain plasma factor level at 50% for further 7 days.
9.6.4 Treatment approach
- Admit to hospital.
- Haemophilia A: give 40 - 50 IU/kg FVIII 12-hourly IV for 1 - 6 days.
- Haemophilia B: give 60 - 80 IU/kg FIX daily for 1 - 6 days.
- Continue maintenance treatment to keep FVIII above 50% and FIX above 30% for 7 - 14 days.
- Concomitant tranexamic acid oral therapy at 1 g 8-hourly should be given.
- Do upper and lower endoscopy and radiological investigations under factor cover to establish site and cause of bleeding.
- Monitor amount of blood loss and transfuse if indicated.
- Involve gastrointestinal and haematological expertise early.
9.7 Genitourinary bleeding
9.7.1 Clinical features
- Gross or microscopic haematuria
- Renal angle tenderness or lower abdominal pain
- May have dysuria or features of urinary tract infection.
9.7.2 Treatment goal
- Raise plasma factor level to 40 - 50% in haemophilia A and 30 - 50% in haemophilia B for 3 - 5 days.
- Ensure that there is no clot formation causing urinary tract obstruction.
9.7.3 Treatment approach
- Do not use tranexamic acid - it is contraindicated in proximal urinary bleeds.
- Give 20 - 25 IU/kg FVIII twice daily for 3 days or 30 -50 IU/kg FIX IV daily for 3 days and then review.
- Increase fluid intake by 2 - 3 litres per day.
- Investigate if haematuria is recurrent or persistent.
- Look for and treat any possible infection with appropriate antibiotics.
9.8 Management of patients undergoing surgery
9.8.1 Types of surgical interventions
- Minor surgery, which includes endoscopy, skin biopsy, bronchoscopy, lumbar puncture, etc.
- Major surgery, which includes laparotomy, arthroplasty.
9.8.2 Preoperative assessment and preparation
- Consultation between surgeon, haematologist and blood centre.
- Check FBC, liver function, renal function and inhibitor level.
- Do factor recovery studies.
- Prepare a written management plan and communicate this to all stakeholders.
9.8.3 Treatment goals
- Raise factor level to 50 - 80% for minor surgery and 80 -100% for major surgery.
- Maintain factor level at 50% for major surgery for at least 7 - 14 days.
- Avoid intraoperative and postoperative blood loss.
9.8.4 Treatment approach
- Haemophilia A: for major surgery, give 40 - 50 IU/kg FVIII and for minor surgery give 20 - 40 IU/kg FVIII, 30 minutes before surgery, 6 hours postoperatively and then 12-hourly thereafter.
- Haemophilia B: for major surgery, give 60 - 80 IU/kg FIX and for minor surgery 20 - 40 IU/kg, 30 minutes before surgery. Repeat the same dose 6 hours postoperatively and then daily thereafter.
- Factor infusion for major surgery should continue for 7 - 14 days. Venous thromboembolism (VTE) prophylaxis using elastic stockings should be considered in all high-risk surgery.
- Keep peak maintenance factor level at 50% until healing has started.
- Introduce postoperative rehabilitation and mobilisation gradually under factor prophylaxis.
- Continuous infusion of factor with a pump may be used.
- Use of antibiotics postoperatively is mandatory.
- Ensure that patient receives adequate analgesia.
10. Management of chronic synovitis and target joints
10.1 General comments
- Chronic synovitis is characterised by synovial hypertrophy with formation of new blood vessels. These vessels are prone to more bleeding, resulting in more synovitis and bleeding.
10.2 Clinical features
- Recurrent joint bleeds not adequately responsive to factor replacement.
- Painless joint swelling.
- Joint will bleed spontaneously.
- X-ray in chronic synovitis may not show joint arthropathy.
- The joint with chronic synovitis then becomes a target joint.
10.3 Treatment goals
- To prevent bleeding by raising plasma factor level above 5% with secondary prophylaxis.
- To break the cycle of bleeding-synovitis-new blood vessel formation-bleeding using surgical or radiological synovectomy.
10.4 Treatment approach
- For a target joint give factor 10 - 20 IU/kg 3 times a week to keep plasma factor VIII or factor IX level above 5%.
- For chronic synovitis, give 40 - 50 IU/kg factor FVIII or 40 - 60 IU/kg FIX before the yttrium synoviorthesis procedure and daily thereafter for 3 days. Intra-articular injection of local anaesthetic and steroids is given at the same time.
11. Management of haemophilia carrier pregnancies
Females who are heterozygous for a haemophilia gene mutation are known as carriers. A heterozygous female can be a carrier of haemophilia without having symptoms, as she has another X-chromosome to produce FVIII and FIX. A wide range of clotting factor levels are seen in carriers, from very low, resembling affected males, to the upper limit of normal.60 Carrier detection and prenatal diagnosis are important so that carriers can make an informed decision on whether or not they will risk having a baby with haemophilia.61 Carriers have a 50% chance of having a son with haemophilia or a daughter who is also a carrier. If there is a family member with haemophilia, DNA studies to identify the gene defect in this person can be used to determine whether the female family member is a carrier.
Knowledge of fetal sex allows invasive testing to be avoided in female pregnancies and enables appropriate management of labour and delivery.62 Case series studies have found that miscarriage and postpartum haemorrhage are more common in haemophilia carriers.63-65
11.1 Management of mother pre-pregnancy
- Provide pre-pregnancy genetic counselling to all carriers.
- Establish carrier status and discuss implication with patient.
- Establish FVIII/FIX gene abnormality if possible.
11.2 On diagnosis of pregnancy
- Take detailed family and personal bleeding history.
- Plan management with obstetrician and haematologist.
- For patients wishing to terminate:
- perform chorionic villi sampling (CVS) at 11 - 13 weeks and do gene testing on males, or
- determine sex of fetus at 14 - 16 weeks. If male, proceed to amniocentesis.
- For patients not wishing to terminate - do nothing.
11.3 During pregnancy
- Measure FVIII/FIX at booking and repeat at 28 and 34 weeks.62
- If factor <50%, it should be replaced if CVS, amniocentesis or termination is needed.
- Formulate and discuss delivery plan with patient and obstetrician.
11.4 During labour and delivery
- Plan for vaginal delivery.
- Avoid scalp monitoring.
- Avoid vacuum delivery.
- Avoid forceps.
- If labour is prolonged, perform caesarean section.
- Take blood from umbilical cord for urgent FVIII/FIX assay.
- Avoid heel pricks for coagulation assays.
- FIX assay can be unreliable in a newborn.
- Give baby oral vitamin K. Avoid intramuscular injection.
11.5 Postpartum
- For FVIII carrier, monitor FVIII level. If the level falls below 50% and the patient is bleeding, give DDAVP or FVIII concentrate.
- For FIX carrier, monitor FIX level and give replacement if the factor level is <50% and the patient is bleeding.
11.6 Management of symptomatic carriers
In females known to be carriers, it is important to assay their FVIII or FIX levels to establish whether they are at increased risk of bleeding. Low clotting factor levels in carriers are associated with menorrhagia, and with prolonged bleeding after tooth extractions, surgery and trauma.60,64 Menorrhagia is a common symptom in carriers with low factor levels, and can cause moderate to severe restrictions in daily life.60 Symptomatic carriers should be managed according to the severity of their symptoms. Treatments include DDAVP (in symptomatic carriers of haemophilia A), tranexamic acid, and clotting factor concentrates. Menorrhagia can be controlled using hormonal (e.g. oral contraceptive), haemostatic or surgical methods.66 Symptomatic carriers should wear appropriate medical identification.67
12. Prophylaxis
12.1 General
- The rationale for prophylaxis is to maintain factor activity above 1% and thus convert a bleeding pattern of severe haemophilia to that of mild/moderate haemophilia.
- Primary prophylaxis is given to infants identified as being at high risk of developing target joints and joint arthropathy.
- Secondary prophylaxis is given when there is a high requirement for on-demand treatment. Regular prophylactic injections will reduce the frequency of bleeding and rebleeding into target joints, and is often used in chronic synovitis.
- Single-dose prophylaxis is injection of a product prior to an event known to cause bleeding.
12.2 Treatment goals of prophylaxis
- To raise factor level above 1% to convert a severe haemophilia bleeding pattern to a mild/moderate pattern.
- To reduce the number of bleeds.
- To prevent or delay development of joint arthropathy.
12.3 Prophylaxis approach
- Haemophilia A: give 25 - 40 IU/kg FVIII 2 - 3 times a week.
- Haemophilia B: give 25 - 40 IU/kg FIX twice a week.
- Prophylaxis increases the amount of factor consumption but reduces the number of bleeds and delays development of joint arthropathy.
- It is still not clear as to when to start prophylaxis, how to start it, what dose to use to start, and when to stop.
13. Management of pain in haemophilia
13.1 Pain aetiology
Pain in people with haemophilia has multiple causes, and these include:
- Joint capsular stretching as a result of haemarthroses
- Haemophilic arthropathy
- Compartment syndrome.
13.2 Goals of pain management
- To remove pain completely without increasing the risk of bleeding.
- To improve the patient's quality of life.
13.3 Pain management principles
- Analgesic agents known to relieve pain without increasing the bleeding risk include Spectrapyn®, Doxyfene®, Tramal™, Nubain® and COX-2 inhibitors.
- COX-2 inhibitors are favoured largely owing to their favourable side-effect profile, analgesic effects, anti-inflammatory effects and anti-angiogenic effects.30
- Aspirin and other antiplatelet agents should be avoided.
- Analgesia requiring intramuscular injection should also be avoided.
14. Conclusion
Much has been achieved for people with haemophilia in South Africa over the last 40 years. In 1968, the first two haemophilia treatment centres were established, at Johannesburg Hospital and Red Cross Children's Hospital in Cape Town. Today there are 16 haemophilia treatment centres in South Africa. These are located in Johannesburg, Cape Town, Port Elizabeth, East London, Durban, Pretoria, Bloemfontein, Potchefstroom, Mthatha and Polokwane, and are responsible for management of over 2 200 people with bleeding disorders in South Africa. More than 80% of these are people with haemophilia.
Data from the WFH have shown that survival to adulthood among people with haemophilia increases fivefold and the cost of their care decreases significantly if they have access to a haemophilia treatment centre.68 In addition to care in such a centre, the WFH has identified two further pivotal elements to achieve maximum impact for minimum input on improving the quality of care delivered to people with haemophilia. These are (i) on-demand treatment with appropriate factor replacement for major bleeding and surgical intervention; and (ii) education of health care providers, patients, and their families about haemophilia.68 This guideline addresses these latter two elements, and forms part of the commitment of the MASAC of the South African Haemophilia Foundation to promote further the welfare of all persons with haemophilia in South Africa.
15. Conflict of interest disclosure
Dr Johnny Mahlangu is the Chairman of MASAC of the South African Haemophilia Foundation and has no conflict of interest disclosure to make. Sister Anne Gilham is Secretary of MASAC and employed by the National Bioproduct Institute of South Africa. Editing and writing assistance was provided by Bioscript Stirling Ltd, UK, which was funded by Novo Nordisk.
16. References
1. Association of Hemophilia Clinic Directors of Canada. Clinical Practice Guidelines. Hemophilia and von Willebrand's Disease: 2. Management. 2nd ed. 1999. http://www.ahcdc.ca/vWDManagement.html (last accessed 1 March 2007). [ Links ]
2. United Kingdom Haemophilia Centre Doctors' Organisation. Guidelines on the selection and use of therapeutic products to treat haemophilia and other hereditary bleeding disorders. Haemophilia 2003; 9: 1-23. [ Links ]
3. World Federation of Hemophilia. Guidelines for the Management of Hemophilia. Montreal: World Federation of Hemophilia, 2005. http://www.wfh.org/2/docs/Publications/Diagnosis_and_Treatment/Gudelines_Mng_Hemophilia.pdf (last accessed 1 March 2007). [ Links ]
4. Medical Advisory Committee of Haemophilia Foundation of New Zealand. National guidelines. Management of haemophilia. Treatment protocols. 2005. http://www.haemophilia.org.nz/uploads/bloodline/TreatmentGUIDELINEFinalrevisedMarch05.pdf (last accessed 5 June 2007). [ Links ]
5. Medical and Scientific Advisory Council of the National Hemophilia Foundation (USA). MASAC recommendations concerning the treatment of hemophilia and other bleeding disorders. 2006. http://www.hemophilia.org/NHFWeb/MainPgs/MainNHF.aspx?menuid=57&contentid=693 (last accessed 5 June 2007). [ Links ]
6. Australian Health Ministers' Advisory Council. Evidence-based Clinical Practice Guidelines for the Use of Recombinant and Plasma-derived FVIII and FIX Products. 2006. http://www.nba.gov.au/PDF/FINAL%20GLs%20with%20bookmarks%20June%202006.pdf (last accessed 1 March 2007). [ Links ]
7. Soucie JM, Evatt B, Jackson D. Occurrence of hemophilia in the United States. The Hemophilia Surveillance System Project Investigators. Am J Hematol 1998; 59: 288-294. [ Links ]
8. Gitschier J, Wood WI, Goralka TM, et al. Characterization of the human factor VIII gene. Nature 1984; 312: 326-330. [ Links ]
9. Lakich D, Kazazian HH Jr, Antonarakis SE, Gitschier J. Inversions disrupting the factor VIII gene are a common cause of severe haemophilia A. Nat Genet 1993; 5: 236-241. [ Links ]
10. Naylor J, Brinke A, Hassock S, Green PM, Giannelli F. Characteristic mRNA abnormality found in half the patients with severe haemophilia A is due to large DNA inversions. Hum Mol Genet 1993; 2: 1773-1778. [ Links ]
11. Antonarakis SE, Rossiter JP, Young M, et al. Factor VIII gene inversions in severe hemophilia A: results of an international consortium study. Blood 1995; 86: 2206-2212. [ Links ]
12. Rossiter JP, Young M, Kimberland ML, et al. Factor VIII gene inversions causing severe hemophilia A originate almost exclusively in male germ cells. Hum Mol Genet 1994; 3: 1035-1039. [ Links ]
13. Bagnall RD, Waseem N, Green PM, Giannelli F. Recurrent inversion breaking intron 1 of the factor VIII gene is a frequent cause of severe hemophilia A. Blood 2002; 99: 168-174. [ Links ]
14. Yoshitake S, Schach BG, Foster DC, Davie EW, Kurachi K. Nucleotide sequence of the gene for human factor IX (antihemophilic factor B). Biochemistry 1985; 24: 3736-3750. [ Links ]
15. Mukherjee S, Mukhopadhyay A, Chaudhuri K, Ray K. Analysis of haemophilia B database and strategies for identification of common point mutations in the factor IX gene. Haemophilia 2003; 9: 187-192. [ Links ]
16. Quadros L, Ghosh K, Shetty S. A common G10430A mutation (Gly 60 Ser) in the factor IX gene describes the presence of moderate and mild hemophilia B in the majority of the Gujarati population. Ann Hematol 2007; 86: 377-379. [ Links ]
17. Attali O, Vinciguerra C, Trzeciak MC, et al. Factor IX gene analysis in 70 unrelated patients with haemophilia B: description of 13 new mutations. Thromb Haemost 1999; 82: 1437-1442. [ Links ]
18. Thorland EC, Weinshenker BG, Liu JZ, et al. Molecular epidemiology of factor IX germline mutations in Mexican Hispanics: pattern of mutation and potential founder effects. Thromb Haemost 1995; 74: 1416-1422. [ Links ]
19. Ketterling RP, Bottema CD, Phillips JA 3rd, Sommer SS. Evidence that descendants of three founders constitute about 25% of hemophilia B in the United States. Genomics 1991; 10: 1093-1096. [ Links ]
20. Hedner U, Ginsburg D, Lusher JM, High KA. Congenital hemorrhagic disorders: new insights into the pathophysiology and treatment of hemophilia. Hematology Am Soc Hematol Educ Program 2000; 2000(1): 241-265. [ Links ]
21. White GC II, Rosendaal F, Aledort LM, Lusher JM, Rothschild C, Ingerslev J. Definitions in hemophilia: recommendation of the scientific subcommittee on factor VIII and factor IX of the scientific and standardization committee of the International Society of Thrombosis and Haemostasis. Thromb Haemost 2001; 85: 560. [ Links ]
22. Stein H, Duthie RB. The pathogenesis of chronic haemophilic arthropathy. J Bone Joint Surg Br 1981; 63B: 601-609. [ Links ]
23. Evatt BL, Black C, Batorova A, Street A, Srivastava A. Comprehensive care for haemophilia around the world. Haemophilia 2004; 10: suppl 4, 9-13. [ Links ]
24. Evatt BL, Austin H, Leon G, Ruiz-Saez A, De Bosch N. Haemophilia therapy: assessing the cumulative risk of HIV exposure by cryoprecipitate. Haemophilia 1999; 5: 295-300. [ Links ]
25. Evatt BL, Farrugia A, Shapiro AD, Wilde JT. Haemophilia 2002: emerging risks of treatment. Haemophilia 2002; 8: 221-229. [ Links ]
26. Lusher JM. Early treatment with recombinant factor VIIa results in greater efficacy with less product. Eur J Haematol Suppl 1998; 63: 7-10. [ Links ]
27. Lusher JM. Acute hemarthroses: the benefits of early versus late treatment with recombinant activated factor VII. Blood Coagul Fibrinolysis 2000; 11: suppl 1, S45-S49. [ Links ]
28. Evans DI, Shaw A. Safety of intramuscular injection of hepatitis B vaccine in haemophiliacs. BMJ 1990; 300: 1694-1695. [ Links ]
29. Goyal R. Intramuscular vaccination in hemophilia. Indian Pediatr 2000; 37: 569. [ Links ]
30. Rattray B, Nugent DJ, Young G. Rofecoxib as adjunctive therapy for haemophilic arthropathy Haemophilia 2005; 11: 240-244. [ Links ]
31. Oon LL, King A, Higgins JA, Lee CA, Kernoff PB, Goodall AH. Protective antibodies to hepatitis B virus in haemophiliacs. J Med Virol 1991; 33: 19-25. [ Links ]
32. Kisker CT, Mahoney EM, Arkin S, Maeder MA, Donfield SM, Evatt BL. Changes in hepatitis B serologic titers in HIV+ and HIV- children with haemophilia. Haemophilia 1999; 5: 354-359. [ Links ]
33. Fischer K, Grobbee DE, van den Berg HM. RCTs and observational studies to determine the effect of prophylaxis in severe haemophilia. Haemophilia 2007; 13: 345-350. [ Links ]
34. Mulder K, Cassis F, Seuser DR, Narayan P, Dalzell R, Poulsen W. Risks and benefits of sports and fitness activities for people with haemophilia. Haemophilia 2004; 10: suppl 4, 161-163. [ Links ]
35. Harris S, Boggio LN. Exercise may decrease further destruction in the adult haemophilic joint. Haemophilia 2006; 12: 237-240. [ Links ]
36. Paton CM, Brandauer J, Weiss EP, et al. Hemostatic response to postprandial lipemia before and after exercise training. J Appl Physiol 2006; 101: 316-321. [ Links ]
37. White GC 2nd, Courter S, Bray GL, Lee M, Gomperts ED. A multicenter study of recombinant factor VIII (Recombinate) in previously treated patients with hemophilia A. The Recombinate Previously Treated Patient Study Group. Thromb Haemost 1997; 77: 660-667. [ Links ]
38. Gouw SC, van der Bom JG, Auerswald G, Ettinghausen CE, Tedgard U, van den Berg HM. Recombinant versus plasma-derived factor VIII products and the development of inhibitors in previously untreated patients with severe hemophilia A: the CANAL cohort study. Blood 2007; 109: 4693-4697. [ Links ]
39. MacGregor IR, Roberts EM, Prowse CV, Broomhead AF, Ozolins M, Litka P. Fibrinolytic and haemostatic responses to desamino-d-arginine vasopressin (DDAVP) administered by intravenous and subcutaneous routes in healthy subjects. Thromb Haemost 1988; 59: 34-39. [ Links ]
40. Mannucci PM, Bettega D, Cattaneo M. Patterns of development of tachyphylaxis in patients with haemophilia and von Willebrand disease after repeated doses of desmopressin (DDAVP). Br J Haematol 1992; 82: 87-93. [ Links ]
41. Lethagen S. Desmopressin in mild hemophilia A: indications, limitations, efficacy, and safety. Semin Thromb Hemost 2003; 29: 101-106. [ Links ]
42. Ruiz-Saez A, Hong A, Arguello A, et al. Pharmacokinetics, thrombogenicity and safety of a double viral inactivated factor IX concentrate compared with a prothrombin complex concentrate. Haemophilia 2005; 11: 583-588. [ Links ]
43. Ehrlich HJ, Henzl MJ, Gomperts ED. Safety of factor VIII inhibitor bypass activity (FEIBA ): 10-year compilation of thrombotic adverse events. Haemophilia 2002; 8: 83-90. [ Links ]
44. Lankiewicz MW, Hays J, Friedman KD, Tinkoff G, Blatt PM. Urgent reversal of warfarin with prothrombin complex concentrate. J Thromb Haemost 2006; 4: 967-970. [ Links ]
45. Preston FE, Laidlaw ST, Sampson B, Kitchen S. Rapid reversal of oral anticoagulation with warfarin by a prothrombin complex concentrate (Beriplex): efficacy and safety in 42 patients. Br J Haematol 2002; 116: 619-624. [ Links ]
46. Kouides PA, Kulzer L. Prophylactic treatment of severe factor X deficiency with prothrombin complex concentrate. Haemophilia 2001; 7: 220-223. [ Links ]
47. McMahon C, Smith J, Goonan C, Byrne M, Smith OP. The role of primary prophylactic factor replacement therapy in children with severe factor X deficiency. Br J Haematol 2002; 119: 789-791. [ Links ]
48. Lossing TS, Kasper CK, Feinstein DI. Detection of factor VIII inhibitors with the partial thromboplastin time. Blood 1977; 49: 793-797. [ Links ]
49. Wight J, Paisley S, Knight C. Immune tolerance induction in patients with haemophilia A with inhibitors: a systematic review. Haemophilia 2003; 9: 436-463. [ Links ]
50. Mauser-Bunschoten EP, Nieuwenhuis HK, Roosendaal G, van den Berg HM. Low-dose immune tolerance induction in hemophilia A patients with inhibitors. Blood 1995; 86: 983-988. [ Links ]
51. Brackmann HH, Oldenburg J, Schwaab R. Immune tolerance for the treatment of factor VIII inhibitors - twenty years' 'Bonn protocol'. Vox Sang 1996; 70: suppl 1, 30-35. [ Links ]
52. Oldenburg J, Schwaab R, Brackmann HH. Induction of immune tolerance in haemophilia A inhibitor patients by the 'Bonn Protocol': predictive parameter for therapy duration and outcome. Vox Sang 1999; 77: suppl 1, 49-54. [ Links ]
53. Freiburghaus C, Berntorp E, Ekman M, Gunnarsson M, Kjellberg B, Nilsson IM. Tolerance induction using the Malmo treatment model 1982-1995. Haemophilia 1999; 5: 32-39. [ Links ]
54. Mathew P. Current opinion on inhibitor treatment options. Semin Hematol 2006; 43 (2): suppl 4, S8-S13. [ Links ]
55. Hay C, Recht M, Carcao M, Reipert B. Current and future approaches to inhibitor management and aversion. Semin Thromb Hemost 2006; 32: suppl 2, 15-21. [ Links ]
56. Chuansumrit A, Isarangkura P, Angchaisuksiri P, et al. Controlling acute bleeding episodes with recombinant factor VIIa in haemophiliacs with inhibitor: continuous infusion and bolus injection. Haemophilia 2000; 6: 61-65. [ Links ]
57. Astermark J, Donfield SM, DiMichele DM, et al. A randomized comparison of bypassing agents in hemophilia complicated by an inhibitor: the FEIBA®, NovoSeven® Comparative (FENOC) Study. Blood 2007; 109: 546-551. [ Links ]
58. Cohen AJ, Kessler CM. Acquired inhibitors. Baillieres Clin Haematol 1996; 9: 331-354. [ Links ]
59. Negrier C, Hay CR. The treatment of bleeding in hemophilic patients with inhibitors with recombinant factor VIIa. Semin Thromb Hemost 2000; 26: 407-412. [ Links ]
60. Plug I, Mauser-Bunschoten EP, Brocker-Vriends AH, et al. Bleeding in carriers of hemophilia. Blood 2006; 108: 52-56. [ Links ]
61. Chandy M. Treatment Options in the Management of Hemophilia in Developing Countries. Montreal: World Federation of Hemophilia, 2005. http://www.wfh.org/2/docs/Publications/Diagnosis_and_Treatment/TOH37_Treatment_Dev_Countries_Add.pdf (last accessed 1 March 2007). [ Links ]
62. Lee CA, Chi C, Pavord SR, et al. UK Haemophilia Centre Doctors' Organization. The obstetric and gynaecological management of women with inherited bleeding disorders - review with guidelines produced by a taskforce of UK Haemophilia Centre Doctors' Organization. Haemophilia 2006; 12: 301-336. [ Links ]
63. Kadir RA, Economides DL, Braithwaite J, Goldman E, Lee CA. The obstetric experience of carriers of haemophilia. Br J Obstet Gynaecol 1997; 104: 803-810. [ Links ]
64. Mauser Bunschoten EP, van Houwelingen JC, et al. Bleeding symptoms in carriers of hemophilia A and B. Thromb Haemost 1988; 59: 349-352. [ Links ]
65. Greer IA, Lowe GD, Walker JJ, Forbes CD. Haemorrhagic problems in obstetrics and gynaecology in patients with congenital coagulopathies. Br J Obstet Gynaecol 1991; 98: 909-918. [ Links ]
66. Siegel JE, Kouides PA. Menorrhagia from a haematologist's point of view. Part II: management. Haemophilia 2002; 8: 339-347. [ Links ]
67. Ross J. Perspectives of Hemophilia Carriers. Revised ed. Montreal: World Federation of Hemophilia, 2004. http://www.wfh.org/2/docs/Publications/Genetic_counsel/TOH-8_English_Carriers.pdf (last accessed 10 April 2007). [ Links ]
68. World Federation of Hemophilia. Fact Sheet 1: Economic Benefits of Basic Hemophilia Treatment & Care. Montreal: World Federation of Hemophilia, 2004. http://www.wfh.org/2/docs/Publications/Hemo_Org_Resources/Fact_sheet_1_Apr04.pdf (last accessed 12 April 2007). [ Links ]
 Correspondence:
Correspondence:
Dr Johnny N Mahlangu
PO Box 76384, Wendywood, 2144, South Africa
Tel.: 011 489 8413, Fax: 011 489 8589
E-mail: johnny. mahlangu@nhls.ac.za


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}