










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSAMJ: South African Medical Journal
On-line version ISSN 2078-5135
Print version ISSN 0256-9574
SAMJ, S. Afr. med. j. vol.98 n.2 Pretoria Feb. 2008
ORIGINAL ARTICLES
Familial hypercholesterolaemia: The Cape Town experience
Jean C FirthI; A David MaraisII
IMB ChB, DCH, PhD; Lipidology Division, Department of Internal Medicine, Groote Schuur Hospital and University of Cape Town
IIMB ChB, FCP (SA); Lipidology Division, Department of Internal Medicine, Groote Schuur Hospital and University of Cape Town
ABSTRACT
Familial hypercholesterolaemia (FH), an autosomal dominantly inherited disorder characterised by elevated plasma low-density lipoprotein (LDL) cholesterol levels, tendon xanthomata and premature ischaemic heart disease, is amenable to treatment with modern medication.
The clinical and biochemical details of 1 031 patients with FH were analysed. FH is the most common monogenic disorder of lipoprotein metabolism presenting to the Lipid Clinic at Groote Schuur Hospital, accounting for about 20% of consultations. The hospital classified 55% of the FH patients as white, 43% as coloured, 1.5% as Asian and 0.5% as black. In the FH cohort (whose mean age at presentation was 44 years), 80% had tendon xanthomata, 36% had arcus cornealis, and 14% had xanthelasma. Tendon xanthomata was present in almost 90% of patients by the age of 50 years. Arcus cornealis was present in about 45% by the age of 40 years, further increasing in frequency with age. Cardiovascular complications included ischaemic heart disease (43%), stroke (1.5%), transient ischaemic attacks (1.3%), and peripheral vascular disease (3.7%). The mean age of death was 55 (±13) years; 51 (±10) years in men and 61 (±12) years in women. In 46% of the cohort, a defective gene was identified by testing for locally prevalent mutations.
Familial hypercholesterolaemia (FH) is a common, serious disease affecting all ethnic groups in South Africa. It can be diagnosed by clinical features and routine laboratory tests at primary health care level. Effective treatment prevents early and debilitating cardiovascular disease.
FH is an autosomal dominantly inherited disorder.1 The heterozygous phenotype is characterised by a personal or family history of premature ischaemic heart disease, tendon xanthomata and elevated plasma low-density lipoprotein (LDL) cholesterol levels (5 - 12 mmol/l). The homozygous phenotype displays tendon and cutaneous xanthomata, higher plasma LDL levels (>15 mmol/l), and often presents in childhood with physical signs or complications of the disease. Although the heterozygous phenotype can be due to at least 3 genes, it most commonly results from mutations in the LDL receptor (LDLR). Mutations in apolipoprotein B, the ligand binding circulating LDL to the LDLR, can produce the same phenotype, as well as mutations in the proconvertase subtilin/ kexin type 9 (PCSK9) gene, which regulates LDLR turnover.
FH was first reported in SA in 1977.2 A higher-than-expected number of subjects with homozygous FH suggested a founder effect in Afrikaners.3 In this group, 3 mutations were identified in the LDLR.4 The prevalence of FH by these genotypes in a rural community was determined to be 1/83,5 compared with a worldwide prevalence estimate of 1/500. Several additional mutations have been described in South Africa6 in various population groups.
We describe here the experience with the first 1 000 patients presenting with the FH phenotype at the Groote Schuur Hospital Lipid Clinic. Clinical and biochemical data were analysed, along with genotyping for founder mutations after exclusion of binding defective apolipoprotein B100.
Methods
Informed consent for research into dyslipidaemia is obtained from all Lipid Clinic patients. Inclusion criteria for the FH phenotype included: an untreated cholesterol level of more than 7.5 mmol/l, attributable to an increase in LDL, together with a tendon xanthoma in the patient (or first-degree relative), and a personal or family history of premature ischaemic heart disease. Where the clinical diagnosis was suspected but data were incomplete, subjects with genetic confirmation were included in the analysis.
An overnight fast was required to determine plasma triglyceride (TG), total cholesterol (TC), high-density lipoprotein cholesterol (HDLC), and LDL cholesterol (LDLC) concentrations. Tests were done with automated analysers using commercial kits. Apolipoprotein AI (apoA-I), apolipoprotein B (apoB) and lipoprotein (a) (Lp(a)) concentrations were also determined. Extraction of DNA7 enabled the identification of the mutations reported in South Africa.8 Dysbetalipoproteinaemia was excluded by non-denaturing gradient acrylamide gel electrophoresis9 and/or by genetic testing for mutations in apolipoprotein E. Agarose gel electrophoresis was performed on all new patients, using a Beckman Paragon system.
Statistical analyses were done with Graphpad Prism and Instat, taking p<0.05 as statistically significant.
Results and discussion
Demographics
Patients on the lipid clinic database totalled 4 494, with more women than men (53% v. 47%). FH constituted the most common monogenic dyslipidaemia (23%), and comprised 55% whites, 43% of persons of mixed ancestry (coloureds), and less than 2% Asians and 1% blacks. New patients over the same time period were distributed differently: whites 40%, coloureds 56%, and Asians and blacks each 2% (Table I).
The prevalence of FH among whites presenting to the clinic is significantly higher than in the coloured group - 55% v. 43% (p<0.0001, chi-squared test). While this may simply reflect the high prevalence of FH due to the founder effect(s) in whites, it may also reflect a greater awareness of this disorder by their medical practitioners, or even a bias that FH is not present in other racial groups.
Estimating the prevalence of FH as being 1/100 for whites, 1/200 in Asians and 1/500 in blacks and coloureds (the numbers of FH subjects calculated according to the 1991 census) for the region within an hour's drive from the Lipid Clinic, led to the figures of 5 700 whites, 100 Asians, 1 000 blacks and 2 300 coloureds. The majority of these cases rely on public health care, requiring referral to the two tertiary hospitals for drug treatment. Assuming that an equal proportion attended each tertiary hospital, it is clear that many adults with FH were not referred, especially in the black population.
In the whole group, males presented younger than females: 72% as opposed to 57% had presented by the age of 50 years (p<0.0001, Fisher's exact test). White males (p=0.0002) and white females (p<0.0001) presented earlier to the clinic than those of mixed ancestry of either sex. The younger presentation is probably due to a greater awareness of FH in the white population and/or among medical practitioners. In contrast, blacks appeared to present latest, often only following a myocardial infarction, this occurring at an average age of 50 years. These findings underscore the need to raise awareness of, and to screen for, FH in the population at large.
Physical signs
In the FH cohort, xanthelasma was uncommon (14%). Arcus cornealis was present in 36%, and 80% had tendon xanthomata. Xanthelasma was significantly commoner in females (p<0.0001, Fisher's exact test). Arcus cornealis was commoner in males (p=0.0009, Fisher's exact test) and in smokers (p<0.0001, Fisher's exact test). The Achilles tendon was abnormal in the majority of cases (79%), but 1% had only extensor tendon xanthomata of the hand.
Physical signs of dyslipidaemia are uncommon before the age of 25 years. There is a marked increase in the prevalence of tendon xanthomata from 12% before the age of 25 years up to 75% by the age of 40 years, and about 90% by the age of 50 years. In both sexes, tendon xanthomata are present in the third decade and well established by the age of 40 years. While the prevalence of tendon xanthomata in males remains static with age, it increases progressively in females, from 76% at the age of 40 to 91% by the age of 70 years (Fig. 1).
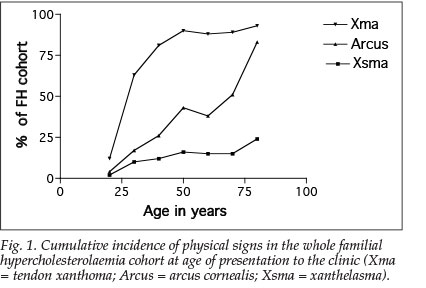
Arcus cornealis is present in about 25% of FH subjects by the age of 40 years and about 45% by the age of 50 years; by the age of 75 years more than 75% of the cohort was affected, possibly due to senescence (arcus senilis). There was no difference in the prevalence of xanthelasma at presentation in males and females (p=0.3), but arcus occurred significantly earlier in males (p=0.0001), as did tendon xanthomata (p<0.0001, chi-squared test).
Referral letters accompanying patients almost invariably commented on xanthelasma, but seldom on arcus cornealis and practically never on tendon xanthomata. This implies that, with the exception of xanthelasma, clinical signs did not present a selection bias in our analysis of physical signs. An Achilles tendon xanthoma is a valuable physical sign for screening for individuals with a high risk of heart disease and to diagnose FH, especially after the age of 40 years.
The association of lipid profiles, including Lp(a), with physical signs was examined (Table II). Patients with xanthelasma had higher fasting plasma TG (p=0.04), TC (p=0.002) and LDLC (p=0.003) (chi-squared test) concentrations than those without xanthelasma. Patients with arcus cornealis had higher TC (p=0.0006) and LDLC (p<0.0001) (chi-squared test) concentrations than those without. In patients with tendon xanthomata, TG (p<0.0001), TC (p=0.006), and LDLC (p=0.015) and Lp(a) (p<0.0001) (chi-squared test) levels were higher than in those without tendon xanthomata. HDLC concentration was significantly lower (p<0.012) in subjects with tendon xanthomata. The pathogenesis of the physical signs is not well understood. Although chronic exposure to high concentrations of LDLC largely determines the development of physical signs, HDL and very low density lipoprotein (VLDL) metabolism may play an additional role in the pathogenesis of tendon xanthomata and xanthelasma, respectively.
Lipid profiles
In untreated fasting lipid profiles the TG concentrations increase with age in both genders (ANOVA, p<0.0001). There was no significant difference in the TC, HDLC or LDLC levels with increasing age, nor in the Lp(a) levels. In concordance with LDLC and HDLC, the apo B and apoA-I concentrations also did not change with age. According to the Fredrickson classification of agarose gel electrophoresis, 80% were type IIa and 20% were type IIb. This suggests that there may be slight increases in VLDL in FH, in response to other common genetic and environmental factors, including the metabolic syndrome (Table III).
Genotype results
All 3 genes causing the heterozygous FH phenotype relate to impaired clearance of LDL and are inherited in an autosomal dominant fashion. The most common cause of FH is the LDL receptor (LDLR) gene, with more than 840 different mutations on record.10
Mutations in apoB may result in a milder phenotype of FH, especially in children,11 but the adult phenotype is generally regarded as indistinguishable from that caused by LDLR mutations.12,13 The identification of 11 index cases with FDB accounted for 1.1% of the FH phenotypes and is too small for statistical comparison. Pro-protein convertase subtillin/kexin type 9 (PCSK9) encodes neural apoptosis regulated convertase 1 (NARC1) and regulates the LDL receptor activity,14 but there is also evidence for over-production of lipoproteins in these patients.15 This disorder was not sought, but is expected to contribute a very small proportion of our patients.
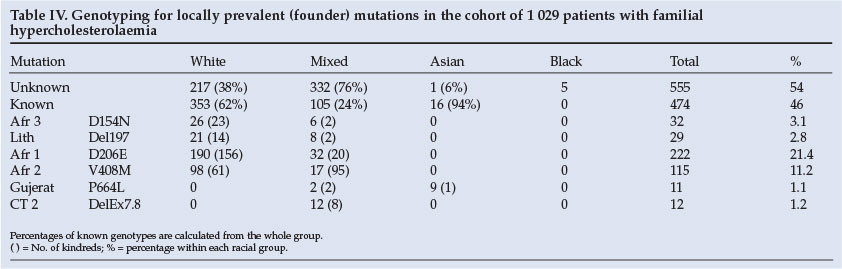
Genetic analysis is summarised in Table IV. All patients were screened for locally prevalent (9) LDLR mutations. The genotyping in whites reflects the founder effects of D154N, D206E and V408M in Afrikaners and del197 in Jews, but these mutations are also present in subjects of mixed ancestry. Founder effects have occurred for E207K and P664L in the Asian (Indian) communities, while none of the tests identified LDLR mutations in blacks.
With this limited diagnostic strategy, approximately 47% of the FH phenotype could be proven by genetic studies. The founder effects permitted 94% identification in Indians and 62% in whites. In accordance with a previous publication,16 76% of white Afrikaners could be identified by their 3 founder genes. In contrast, only 24% of FH could be identified in the mixed ancestry population where the Afrikaner mutations predominated. This agrees with a previous analysis of this cohort in the clinic.17 The 2.5 kb deletion of exons 7 and 8 (Cape Town 2) mutation appears to be exclusive to the mixed ancestry group.
Complications
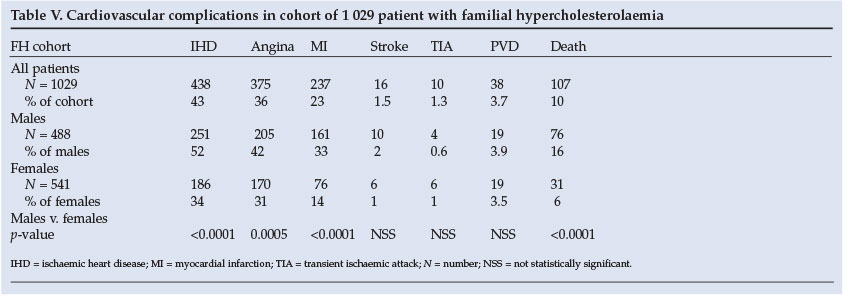
Cardiovascular complications are displayed in Table V. Cardiac complications, including death, occurred more frequently in males than in females, but still affected more than a third of females. Other vascular complications were uncommon and occurred equally in both sexes.
Only 43% of the FH cohort had ischaemic heart disease (IHD), at presentation and during follow-up, with males predominating (52% v. 34%, p=0.0005, Fisher's exact test). Of those developing IHD, 68% were smokers, with a higher percentage in males (78% v. 55%, p<0.0001, Fisher's exact test). Hypertension was present in 24% with IHD, with more female hypertensives (33% v.16%, p<0.0001, Fisher's exact test).
Stroke was remarkably uncommon in the FH cohort (1.6%) and had a strong association with smoking (94%) and hypertension (63%), but there was no association with body mass index, waist/hip ratio, HDLC, LDLC and Lp(a).
Peripheral vascular disease (PVD) was uncommon (<4%). The prevalence of smoking in patients with PVD was high (79%), especially in males (95% v. 63%). About a quarter of the FH cohort with PVD was diabetic. Diabetes was present in about 5% of the entire FH cohort and occurred similarly in males and females.
The average age of death in the FH cohort followed at the clinic was 54.9 (±13) years, with that of men being 51.4 (±10) and women 60.5 (±12) years. More males died during follow-up, and their age at death was 10 years younger than that for females, although this was not statistically significant (p=0.09, Fisher's exact test).
We compared cardiovascular disease in the first 122 genotyped FH patients in 1994 with the current analysis. There was no significant difference in the average age of onset of angina pectoris (46 ± 11 years) or myocardial infarction (45 ± 11 years). This may reflect a failure to diagnose FH and thus a failure to undertake preventive action. In contrast, there was a significant delay in the average age of death, from 48 years to 55 years (p=0.012, unpaired t-test). The improvement in survival is probably due to treatment with statins. LDLC levels were generally lowered by about 30% to 40% with 20 mg of simvastatin given at night. In addition it is likely that aggressive lifestyle management, especially lowering of cholesterol and saturated fat intake and cessation of smoking, played significant roles.
Conclusions
FH is a common heritable disorder which can be diagnosed clinically at primary health care level. A strong predisposition to premature coronary artery disease (CAD) can be modulated by lifestyle and drug treatment. Although FH is most commonly encountered in Afrikaners, it occurs in all communities and is under-diagnosed. Simple screening of TC by a finger-prick test and a good family history and physical examination would identify most FH individuals. A raised TC (>7.5 mmol/l) or LDLC (>5 mmol/l) level in the presence of normal TG suggests the diagnosis and is confirmed by presence of a tendon xanthoma.
Genetic confirmation of the diagnosis of FH is not always successful. As a result of founder effects, 76% of the Afrikaners were confirmed to have LDLR mutations, whereas only 25% in the coloured group could be confirmed genetically, using a limited strategy for genotyping. In the Jewish patients, 65% had FH Lithuania, and 2 mutations accounted for 94% of FH in the Indian patients.
A diet low in cholesterol and saturated fat lowered the TC by 1.5 mmol/l (data not shown), and should be started during childhood. Statin doses should be titrated to achieve a target LDLC level of 2.5 mmol/l in secondary prevention and in lower-risk subjects, to 3 mmol/l. Individuals vary considerably in response, and monotherapy may not be sufficient. Experienced practitioners should undertake combination treatment. The age to commence drug therapy is not clearly established and may be influenced by cardiovascular risk factors such as hypertension, family history of very premature IHD, and the concentrations of LDLC, HDLC and Lp(a). In general, men should be treated from their early twenties and women later, especially if they plan to have children early in their reproductive years.
Lipid clinics can efficiently diagnose and equitably treat FH while providing valuable clinical, biochemical and genotypic information. This approach is valuable for determining factors that modulate the natural history as well as the other genes responsible for the FH phenotype of this severe disorder. Owing to the high prevalence of FH, patients should continue treatment at primary and secondary health care services. Family members should be investigated.
We thank the Provincial Government of the Western Cape for access to patient records; the contribution of the MRC Cape Heart Group to funding; colleagues in the Lipid Clinic (Dr M Bateman, Dr D Blom, Dr K Wolmarans and Sister J Ross); and the laboratory staff of the Lipid Laboratory (Ms P Byrnes, Ms S Jones, Sister R Jooste, Mrs M Moodie and Ms B Ratanjee).
References
1. Marais AD. Familial hypercholesterolaemia. Clin Biochem Review 2004; 25: 49. [ Links ]
2. Stein EA. The Lipid Disorders Centre at the Transvaal Memorial Hospital for Children: a review of the first thirty months. S Afr Med J 1977; 52: 573-579. [ Links ]
3. Seftel HC, Baker S, Sandler MP, et al. A host of hypercholesterolaemic homozygotes in South Africa. BMJ 1980; 281: 633-636. [ Links ]
4. Kotze M, Langenhoven E, Warnich L, et al. The identification of two low-density lipoprotein receptor gene mutations in South African familial hyoerchoalsterolaemia. S Afr Med J 1989; 76: 399-401. [ Links ]
5. Steyn K, Goldberg YP, Kotze MJ, et al. Estimation of the prevalence of familial hypercholesterolemia in a rural Afrikaner community by direct screeningfor three Afrikaner founder low density lipoprotein receptor gene mutations. Hum Genet 1996; 98(4): 479-484. [ Links ]
6. Vergotine J, Thiart R, Kotze M. Clinical versus molecular diagnosis of heterozygous familial hypercholesterolaemia in diverse South African population. S Afr Med J 2001; 91: 1053-1059. [ Links ]
7. Parzer S, Mannhalter C. A rapid method for the isolation of genomic DNA from citrated whole blood. Biochem J 1991; 273(Pt 1): 229-231. [ Links ]
8. Rubinzstein D, Coetzee GA, Marais AD, Leitersdorf E, Seftel HC, van der Westerhuizen DR. Identification and properties of the proline664-leucine mutant LDL receptor in South Africans of Indian origin. J Lipid Res 1992; 33(11): 1647-1655. [ Links ]
9. Blom DJ, Byrnes P, Jones S, Marais AD. Non-denaturing polyacrylamide gradient gel electrophoresis for the diagnosis of dysbetalipoproteinemia. J Lipid Res 2003; 44(1): 212-217. [ Links ]
10. Villeger L, Abifadel M, Allard D, et al. The UMD-LDLR Database. Hum Mutat 2002; 20: 81-87. [ Links ]
11. Pimstone SN, Defesche JC, Clee SM, Bakker HD, Hayden MR, Kastelein JJ. Differences in the phenotype between children with familial defective apolipoprotein B-100 and familial hypercholesterolemia. Arterioscler Thromb Vasc Biol 1997; 17(5): 826-833. [ Links ]
12. Defesche JC, Pricker KL, Hayden MR, van der Ende BE, Kastelein JJ. Familial defective apolipoprotein B-100 is clinically indistinguishable from familial hypercholesterolemia. Arch Intern Med 1993; 153(20): 2349-2356. [ Links ]
13. Myant NB. Familial defective apolipoprotein B-100: a review, including some comparisons with familial hypercholesterolaemia. Atherosclerosis 1993; 104(1-2): 1-18. [ Links ]
14. Abifadel M, Varret M, Rabes JP, Allard D, Ougerram K, Devilliers Meal. Mutations in PCSK9 cause autosomal dominant hypercholesterolaemia. Nat Genet 2003; 34: 154-156. [ Links ]
15. Sun XM, Eden ER, Tosi I, et al. Evidence for effect of mutant PCSK9 on apolipoprotein B secretion as the cause of unusually severe dominant hypercholesterolaemia. Hum Mol Genet 2005; 14(9): 1161-1169. [ Links ]
16. Graadt vR, Van der Westhuyzen DR, Marais AD, Gevers W, Coetzee GA. Low density lipoprotein receptor founder mutations in Afrikaner familial hypercholesterolaemic patients: a comparison of two geographical areas. Hum Genet 1991; 88(2): 204-208. [ Links ]
17. Loubser O, Marais AD, Kotze M, et al. Founder mutations in the LDL receptor gene contributes significantly to the familial hypercholersterolaemia phenotype in the indigenous South African population of mixed ancestry. Clin Genet 1999; 55: 340-345. [ Links ]
Accepted 5 November 2007.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}