










Serviços Personalizados
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkSouth African Journal of Surgery
versão On-line ISSN 2078-5151
versão impressa ISSN 0038-2361
S. Afr. j. surg. vol.56 no.2 Cape Town Jun. 2018
http://dx.doi.org/10.17159/2078-5151/2018/v56n2a2391
CASE REPORT
Malignant paraganglioma in an African patient associated with a succinate dehydrogenase subunit B (SDHB) mutation
Dennis ShoneI; Jacqueline GoedhalsII; Nicholas E PearceI
IDepartment of Surgery, Faculty of Health Sciences, University of the Free State, Bloemfontein, South Africa
IIDepartment of Anatomical Pathology, National Health Laboratory Service and Faculty of Health Sciences, University of the Free State, Bloemfontein, South Africa
ABSTRACT
We report a case of a malignant extra-adrenal paraganglioma with an underlying succinate dehydrogenase subunit B (SDHB) mutation in an African patient. The focus of this case is the lack of information regarding the underlying susceptibility genes in patients with paragangliomas and pheochromocytomas in Africa.
Introduction
Pheochromocytomas (PCCs) and paragangliomas (PGLs) are rare tumours arising from chromaffin tissues. Pheochromocytomas occur in the adrenal medulla, while paragangliomas occur in extra-adrenal locations and can be divided into sympathetic and parasympathetic categories.1
Approximately 70% of PCCs/PGLs have an underlying genetic abnormality. Currently more than 20 susceptibility genes have been identified,2 including RET, VHL, NF1, TMEM127, KIFlß, MAX, EPAS1, four succinate dehydrogenase subunits (SHDA, SDHB, SDHC, SDHD) and a succinate dehydrogenase (SDH) assembly protein, SDHAF2.2-4 These mutations can be either germline or somatic. Germline mutations occur in germ cells and are hereditary, while somatic mutations cannot be passed on to offspring. This has important implications with regard to genetic counselling, as family members of patients with tumours with germline mutations require screening, while those with somatic mutations do not. Curras-Freixes et al. found germline mutations in 28.7% of PGLs and 4.5% of PCCs.2 The Endocrine Society Clinical Practice Guideline recommends the use of a clinical feature-driven diagnostic algorithm to determine which patients should undergo genetic testing.5 Targeted genetic testing should be provided to patients with a clinical syndrome, while patients with metastatic PGLs should initially be tested for SDHB mutations followed by SDHD, SDHC, VHL and MAX. Patients presenting at a young age, with a positive family history or with multifocal PGLs or bilateral PCCs should also be sent for genetic testing.5
To the best of our knowledge, this is the first reported case of an SDHB-associated paraganglioma from Africa. Approval to report this case was obtained from the Health Sciences Research Ethics Committee of the Faculty of Health Sciences, University of the Free State (ECUFS 73/2016) and the Free State Province Department of Health.
Case report
A 23-year-old self-reported African male with an abdominal mass and secondary hypertension was referred to our tertiary hospital for further surgical assessment. His main complaints were palpitations, feelings of impending doom and unexplained weight loss. Further medical history revealed that he had been treated for uncontrolled hypertension for the past 2 months. Both his father and an uncle on his father's side had died at less than 35 years of age from complications secondary to hypertension. No postmortem examinations had been conducted and the cause of the hypertension had not been investigated in these two relatives.
On examination, he had a pulse rate of 105 beats per minute and a blood pressure of 160/110 mmHg, which remained high over the next 3 days. On abdominal examination, he had a large epigastric mass which was firm, non-tender and immobile. The rest of his examination was normal.
The patient underwent extensive special investigations to determine the underlying cause. The 24-hour urine normetanephrine level was 141 881 nmol/24 hours. A CT scan showed a 9.1 x 10.2 x 6.3 cm retroperitoneal mass that stretched from the level of the aorta bifurcation into the porta. There was abutment of both his renal arteries, superior mesenteric artery and the abdominal aorta. The CT scan also showed lytic lesions involving the right sacroiliac joint and T12 vertebral body suggestive of metastatic disease. All other investigations were normal.
A diagnosis of a malignant extra-adrenal paraganglioma was made. The patient was prepared pre-operatively with crystalloid volume expansion and started on doxazosin, a selective alpha 1 blocker, and amlodipine, a calcium channel blocker. Once the patient was alpha blockaded (defined as experiencing orthostatic hypotension), he was taken to theatre.
An explorative laparotomy was performed. The tumour was found to extend from below the aorta bifurcation up to the third part of the duodenum with infiltration of the serosa of the duodenum. Laterally it abutted both renal arteries and the inferior vena cava. The tumour was macroscopically resected, together with the third part of the duodenum, which was reconstructed with an end-to-side duodeno-enterostomy.
No hypertensive episodes occurred during manipulation of the tumour prior to removal. Once the tumour was removed, the patient developed hypotension, for which adrenaline and phenylephrine were administered. Post-operatively the patient was taken to a surgical intensive care unit and kept intubated until the following day. The duration of the procedure was 380 minutes with a total blood loss of 1 200 mL.
Following surgery, the patient underwent a whole body MIBG and SPECT/CT, which confirmed multiple skeletal metastases. Histological examination of the resection specimen demonstrated a paraganglioma which had invaded the wall of the small bowel. No tumour was noted in the lymph nodes, but there was tumour present on the surgical margins. The patient's symptoms and hypertension, however, resolved.
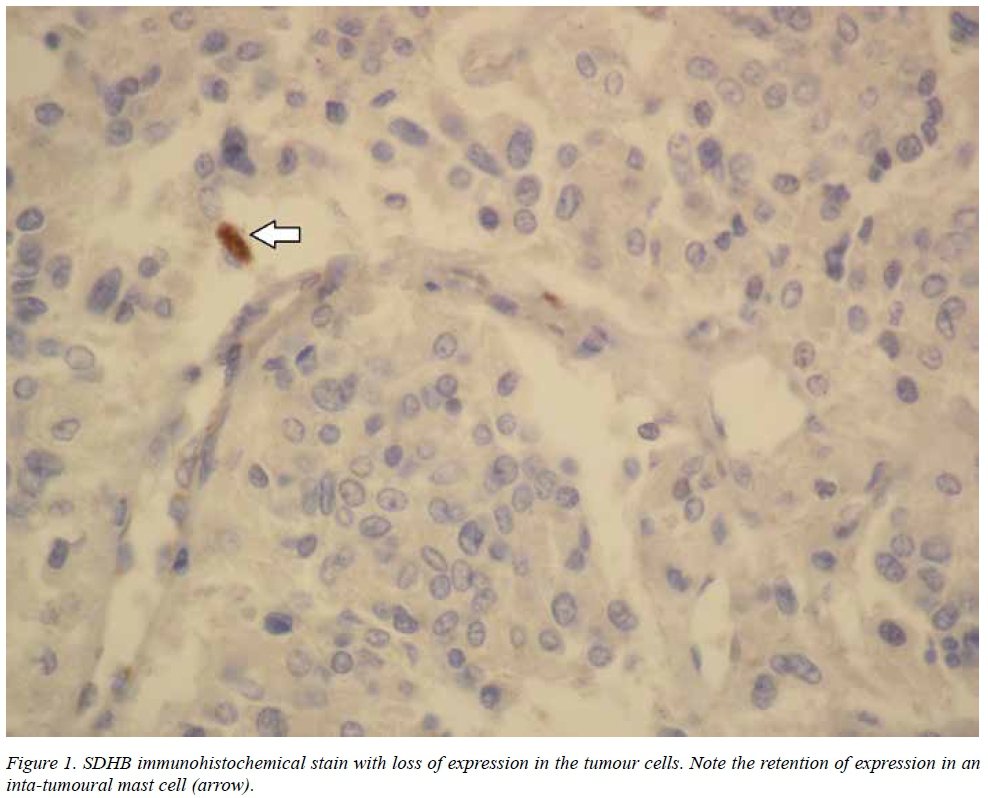
Taking into account the patient's age, family history, the size of the tumour and the presence of metastases, the specimen was referred to the Department of Pathology at Brigham and Women's Hospital, Harvard Medical School in Boston, Massachusetts, for immunohistochemistry for succinate dehydrogenase subunit proteins, as this was not available in South Africa at the time. The tumour cells showed loss of expression of SDHB (Figure 1) and retention of SDHA expression suggesting an underlying SDHB, SDHC or SDHD mutation. Sanger sequencing confirmed a novel SDHB mutation. The patient received genetic counselling and was then referred to the Department of Nuclear Medicine for 131I-MIBG therapy. His family has also undergone genetic counselling and clinical evaluation, but no additional family members with paragangliomas have been identified yet.
Discussion
Most PCCs/PGLs are benign. However, up to 15% of PCCs and 50% of PGLs are malignant.4 Although there are a number of scoring systems available to help predict the risk of metastases, they are not always reliable. The World Health Organization's (WHO) definition of malignancy is the presence of distant metastases at sites where chromaffin tissue does not normally occur. Sites most commonly involved include lymph nodes, bone, liver and lung.4
Treatment options for patients with malignant PCCs/ PGLs include perioperative blood pressure control, surgical removal or debulking, 131I-MIBG and systemic chemotherapy regimens. Recently targeted therapies, such as tyrosine kinase inhibitors, are being explored, but data are still limited.1 Despite all these therapies, the 5-year survival remains poor.1
Germline mutations in the SDHB gene are known to increase the risk of malignancy.3 With most population-based studies regarding susceptibility genes coming from Europe and the United States, no literature on the occurrence of specific genetic abnormalities in PGLs/PCCs in the African population is currently available. Although studies on pheochromocytomas have been reported,6 no relevant studies on the incidence of paragangliomas in Africa could be located in the literature.
The percentage of tumours with an underlying mutation and the profile of these genetic abnormalities in this population are therefore unknown and require further investigation. Identification of these susceptibility genes would allow clinicians to manage these patients more appropriately with more accurate prognostic information. It would also identify those patients whose families should be sent for genetic counselling and testing. Any family members found to have mutations should then undergo regular tumour surveillance.
Immunohistochemistry for SDHA and SDHB has been added to our diagnostic platform and is now performed routinely on all PGLs and PCCs received. In addition, a project is currently underway in which next generation sequencing will be performed on 96 cases of PCCs/PGLs to determine the profile of mutations in the South African population.
Acknowledgements
Dr. Daleen Struwig, medical writer/editor, Faculty of Health Sciences, University of the Free State, for technical and editorial preparation of the manuscript.
REFERENCES
1. Van Hulsteijn LT, Niemeijer ND, Dekkers OM, Corssmit, EPM. 131I-MIBG therapy for malignant paraganglioma and phaeochromocytoma: systemic review and meta-analysis. Clin Endocrinol. 2014;80(4):487-501. Available from: http://dx.doi.org/10.1111/cen.12341 [ Links ]
2. Currás-Freixes M, Inglada-Pérez L, Mancikova V, et al. Recommendations for somatic and germline genetic testing of single pheochromocytoma and paraganglioma based on findings from a series of 329 patients. J Med Genet. 2015;52(10):647-56. Available from: http://dx.doi.org/10.1136/jmedgenet-2015-103218. PMID 26269449. [ Links ]
3. Menara M, Oudijk L, Badoual C, et al. SDHD immunohistochemistry: a new tool to validate SDHx mutations in pheochromocytoma/paraganglioma. J Clin Endocrinol Metab. 2015;100(2):287-91. Available from: http://dx.doi.org/10.1210/jc.2014-1870. PMID 25405498. [ Links ]
4. Fishbein L, Orlowski R, Cohen D. Pheochromocytoma/ paraganglioma: review of perioperative management of blood pressure and update on genetic mutations associated with pheochromocytoma. J Clin Hypertens. 2013;15(6):428-34. Available from: http://dx.doi.org/10.1111/jch.12084. PMID 23730992. [ Links ]
5. Lenders JWM, Duh QY, Eisenhofer G, et al. Pheochromocytoma and paraganglioma: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2014;99(6):1915-42. Available from: http://dx.doi.org/10.1210/jc.2014-1498. PMID 24893135. [ Links ]
6. Huddle KRL. Pheochromocytoma in black South Africans - a 30-year audit. S Afr Med J. 2011;101(3):184-8. Available from: http://dx.doi.org/10.7196/SAMJ.4320. PMID 21382250. [ Links ]
 Correspondence:
Correspondence:
Dr. Dennis Shone
dennisshone@gmail.com


{kind=link}