










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSouth African Journal of Surgery
On-line version ISSN 2078-5151
Print version ISSN 0038-2361
S. Afr. j. surg. vol.51 n.1 Cape Town Jan. 2013
http://dx.doi.org/10.7196/SAJS.1314
GENERAL SURGERY
DOI:10.7196/SAJS.1314
Mismatch repair deficiency in colorectal cancer patients in a low-incidence area
F VergouweI; A BoutallII; D StupartIII; U AlgarIV; D GovenderV; G D van der LindeVI; A MallVII; R RamesarVIII; P A GoldbergIX
IMSc. Colorectal Unit, Department of Surgery, University of Cape Town and Groote Schuur Hospital, Cape Town
IIMB ChB, FCS (SA), Cert Gastroenterol (Surg). Colorectal Unit, Department of Surgery, University of Cape Town and Groote Schuur Hospital, Cape Town
IIIMB ChB, FCS (SA). Colorectal Unit, Department of Surgery, University of Cape Town and Groote Schuur Hospital, Cape Town
IVMSc Nursing. Colorectal Unit, Department of Surgery, University of Cape Town and Groote Schuur Hospital, Cape Town
VMB ChB, FCS (SA), Cert Gastroenterol (Surg). Colorectal Unit, Department of Surgery, University of Cape Town and Groote Schuur Hospital, Cape Town
VIMB ChB, FCPath (SA), FRCPath, MMed, PhD. Division of Anatomical Pathology, University of Cape Town, and National Health Laboratory Service, Groote Schuur Hospital, Cape Tow
VIIMB ChB, MMed (Anat Path). National Health Laboratory Services, Kimberley, Northern Cape
VIIIPhD. Surgical Laboratory, University of Cape Town and Groote Schuur Hospital, Cape Tow
IXMSc, PhD. MRC/UCT Human Genetics Research Unit, Division of Human Genetics, and Institute for Infectious Diseases and Molecular Medicine, University of Cape Town and Groote Schuur Hospital, Cape Town
ABSTRACT
BACKGROUND: In a previous study we identified 206 patients with colorectal adenocarcinoma in the Northern Cape province of South Africa, diagnosed between January 2002 and February 2009. The age-standardised incidence was 4.2/100 000 per year world standard population. This is 10% of the rate reported in First-World countries. In high-incidence areas, the rate of abnormal mismatch repair gene expression in colorectal cancers is 2 - 7%.
OBJECTIVES: The aim of this study was to determine the prevalence of hMLH1- and hMSH2-deficient colorectal cancer in the Northern Cape.
METHODS: Formalin-fixed paraffin wax-embedded tissue blocks from 87 colorectal adenocarcinomas identified in the previous study were retrieved. Standard immunohistochemical staining methods were used to detect the expression of hMLH1 and hMSH2 (i.e. products of the hMLH1 and hMSH2 genes) in the tumours using heat-induced antigen retrieval and diaminobenzidene as a chromogen.
RESULTS: In 8 blocks there was insufficient tumour tissue and in 1 case the immunohistochemical staining failed, probably owing to poor fixation, leaving 78 cases for analysis. In 11 cases hMLH1 was deficient and in 6 cases hMSH2 was deficient. Overall, 21.8% of cancers were deficient for hMLH1 or hMSH2.
CONCLUSION: Presuming that 80% of all hMLH1 deficiencies are due to hypermethylation of the gene, we found 10.5% of colorectal cancers in an area with a low incidence of colorectal cancer to be deficient in the product of the mismatch repair gene/s. This is approximately three times the reported rate in high-incidence areas.
The incidence of colorectal cancer (CRC) varies widely throughout the world.[1] Sub-Saharan countries, including South Africa, are reported to be areas with a low incidence of CRC compared with Western countries.[1-3] CRC in the Northern Cape province of South Africa is uncommon, a recent study reporting the age-standardised rate to be 4.2/100 000[4] and the incidence to be one- tenth that in the Western world.[3,5,6]
CRCs can be divided into two initial categories: those that occur sporadically and those with evidence of heritability. The latter category includes cases where the heritability is an obvious Mendelian pattern of dominant inheritance and those where the family merely has a higher than usual incidence of CRC. In economically developed countries, these three clinical patterns of occurrence account for approximately 70 - 85%, 10 - 20% and 5 - 10%, respectively, of all cases of CRC.[7-12] Figures for developing countries are still unknown, but it is likely that their incidence of inherited CRC is similar to that of developed countries.
The most commonly inherited CRCs are familial adenomatous polyposis (FAP) and hereditary non-polyposis colon cancer (HNPCC) or Lynch syndrome (LS, as it is referred to when a disease-causing mutation is identified in the DNA mismatch repair genes). These syndromes are both the result of specific germline mutations. FAP accounts for less than 1% of CRCs. LS is the most common form of inherited CRC, accounting for 2 - 7% of all cases.[9,11,12]
There are a number of methods that can be used to make the clinical diagnosis of LS: analysis of family history using Amsterdam or Bethesda criteria;[13] tumour testing with immunohistochemistry (IHC) detecting the loss of DNA mismatch repair (MMR) gene product; tumour testing for microsatellite instability (MSI); or direct DNA genetic testing. Tumour IHC is a cheap and highly sensitive and specific strategy to screen for LS.[14-17]
An identified LS cohort in the Northern Cape has more than 100 individuals who undergo colonoscopy annually. The contribution of this group to the overall burden of CRC in a low-incidence area is unknown. We hypothesise that in a low CRC incidence area, inherited cancers form a greater proportion of the overall disease burden. The aim of this study was to determine the prevalence of LS by testing for deficiency of hMLH1 and hMSH2 (products of the hMLHl and hMSH2 genes) with IHC in CRCs in the Northern Cape. This report is the first to attempt to provide prevalence data on inherited CRC in a low-incidence area.
Materials and methods
Ethics
Ethical approval was obtained from the Research Ethics Committee of the University of Cape Town, project reference no. 129/2010.
Data collection and specimen retrieval
In a previous study, 206 patients with adenocarcinoma of the colon or rectum diagnosed between 2002 and 2009 in the Northern Cape were identified.[4] The formalin-fixed paraffin blocks of tumours of these patients were retrieved from the National Health Laboratory Service (NHLS) in Kimberley (Northern Cape) and Cape Town (Western Cape). At least 5 tissue sections of each specimen, with a thickness of 3 - 4 µηι, were cut on a microtome (Accu-Cut SRM 200 Rotary Microtome, Sakura Finetek, Torrance, CA, USA) and put on individual silane-coated slides (Marienfeld Laboratory Glassware, Lauda-Königshofen, Germany). All sections were heat-fixed on the slides at 65°C in an incubator (Lab Aire or Labaire Oven) for 30 minutes.
Histochemistry
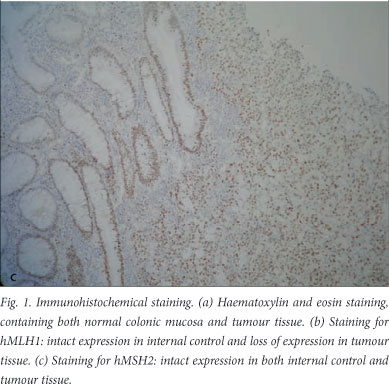
On one section of each case, haematoxylin and eosin staining[18] was performed to make sure that all slides contained both normal colonic mucosa and tumour tissue on the same slide (Fig. 1, a). After dewaxing the sections in xylol for 3x3 minutes, sections were rehydrated through graded alcohols to water for 1 minute each. The series of alcohols consisted of three absolute alcohols, two 96% alcohols and one 70% alcohol. Sections were counterstained in Mayer's haematoxylin[18] for 9 minutes, destained in acid alcohol for 10 seconds, blued in Scott's tap water substitute for 2 minutes, counterstained in eosin for 2 minutes, and dehydrated through graded alcohols to xylol. All slides were mounted in Entellan (Merck KGaA, Darmstadt, Germany).


Immunohistochemical analysis Rationale
LS is an autosomal dominant inherited syndrome caused by germline mutations in one of the DNA-MMR genes, including hMSH2, hMLH1, hMSH6 and hPMS2. hMLH1 and hMSH2 are mutated in about 80 - 90% of all LS cases.[9,10,19] The deficiency of properly working MMR genes results in MSI. Microsatellites are repetitive DNA sequences of 1 - 4 base nucleotides that are particularly sensitive to DNA replication errors when the MMR system is absent.[20] Failure of the MMR system can ultimately lead to the occurrence of multiple colonic and extra-colonic tumours at an early age of onset. Tumour IHC testing for MMR proteins (hMLH1, hMSH2, hMSH6 and hPMS2) is an accurate marker for MMR gene product deficiency.[14,21] Absence of the product of one of these genes in a tumour would indicate that the tumour follows a MMR gene mutation pathway and might be inherited. Tumour IHC is a highly sensitive and specific strategy to screen for LS.[14-17]
Method
After dewaxing in xylol and rehydrating through graded alcohols to tap water, endogenous peroxidase activity was blocked using 1% hydrogen peroxide (Cameron Chemical Consultants, Cape Town, South Africa) solution in methanol for 15 minutes. The slides were rinsed for 5 minutes in running tap water and antigenic sites were unmasked by heat-mediated antigen retrieval for 1.5 minutes at full pressure in disodium ethylenediaminetetra-acetate (EDTA) buffer, pH 8.0 (Protea Laboratory Services, Johannesburg, South Africa), by means of a Presto pressure cooker (Amalgamated Appliances Holdings, Reuven, South Africa). After cooling down in running tap water for 10 minutes, the slides were rinsed with phosphate-buffered saline-Tween (PBST) (Merck, München, Germany). Normal goat serum in phosphate buffer (Dako Denmark A/S), in a concentration of 1:20, was applied for 10 minutes. The slides were drained and the respective mouse antibody was applied immediately at room temperature for 60 minutes. The purified Mouse Anti-Human MLH-1, clone G168-15 (Pharmingen, San Diego, CA, USA), was used at a final dilution of
1/100 and Anti-MSH2 Mouse mAb, clone FE11 (Calbiochem), at a final dilution of 1/500. After rinsing with PBST, the secondary antibody Dako Envision+ System-HRP Labelled Polymer Anti mouse (Dako, Carpinteria, CA, USA) was applied for 30 minutes. Positivity was developed by applying the chromogenic substrate Dako Liquid DAB+ Substrate Chromogen System (Dako North America) for 8 minutes. Slides were rinsed with PBST and tap water and immersed in 1% copper sulphate for 10 minutes to enhance the DAB. Sections were lightly counterstained in Mayer's haematoxylin[18] for 1 minute, blued in Scott's tap water substitute for 2 minutes and dehydrated through graded alcohols to xylol. All slides were mounted in Entellan (Merck KGaA, Darmstadt, Germany).
Normal appendix and normal colonic mucosa were used as positive (intact expression) external controls for the assay. The negative (loss of expression) external control omitted the primary antibody and substituted PBST. Internal control for each patient's slide was the normal colonic mucosa adjacent to the tumour.
The immunohistochemical analysis was done according to CAP (College of American Pathologists) guidelines.[22]
Patient and tumour characteristics
In a previous study, pathology reports of all colorectal adenocarcinomas were retrieved from the NHLS in Kimberley and Cape Town. For this study, wherever possible, patient and tumour characteristics were obtained.[4]
Statistical analyses
Statistical analysis was performed using the STATA/IC 11 program, and a chi-square test was utilised. For the characteristic synchronous lesion, where there were cell counts of <5, Fisher's exact test was performed. A p-value of less than 0.05 was considered statistically significant.
Results
In this study of the original 206 cases, only 85 formalin-fixed paraffin blocks of tumours, together with the pathology reports, were retrieved from the NHLS in Kimberley and 2 blocks from their laboratories in Cape Town, resulting in a total of 87 cases. We excluded 119 cases because we were unable to retrieve the blocks or the pathology reports or could not obtain enough tumour tissue from the blocks.
In 8 of the 87 retrieved blocks, the sections did not contain sufficient tumour tissue. In 1 case the tumour was deficient for both hMLH1 and hMSH2, meaning that the immunohistochemical staining failed (i.e. the inbuilt control was negative); 78 blocks were therefore successfully analysed, of which 45 were biopsies and 33 resection specimens (Fig. 2).

MMR gene product status
After immunohistochemical staining, 17 of the 78 tumours (21.8%) were deficient for hMLH1 or hMSH2; 11 were deficient for hMLH1 (Fig. 1, b) and in 6 cases hMSH2 product was absent. Intact expression of hMSH2 is shown in Fig. 1, c. Clinical and pathological features of specimens deficient for hMLH1 or hMSH2 are summarised in Table 1.
Age/gender
Ages were not available for 2 of the 78 patients. The median age at diagnosis of adenocarcinoma was 58.5 years (standard deviation (SD) 15 years). There was no significant difference in mean age between the two groups (56.9 years in gene product-deficient tumours v. 58.9 years in tumours in which gene product was present). Of the 76 tumours, 20 (26.3%) were diagnosed in patients under the age of 50 years, of which 7 (9.2% of the total) were deficient for gene product on immunohistochemical staining. However, this was not statistically significant (p=0.114). Of the patients, 34 (42.6%) were men and 44 (56.4%) women. There was no difference in gene product status by gender.
Histological features
The majority of the CRCs were regular adenocarcinomas, but in 8 cases (10.3%) the tumour was a mucinous or signet ring cell carcinoma. There was a significant difference (p=0.041) in MMR status by histological type, with more mucinous and signet ring cell carcinomas among the MMR-deficient tumours (23.5% v. 6.6%).
Differentiation
The grade of differentiation was identified in 67 cases: 59 tumours (88.1%) were moderately or well differentiated and 8 (11.9%) were poorly differentiated. The MMR-deficient tumours were more often poorly differentiated (p=0.002).
Site
The site of the CRC could be identified from the pathology report in 71 cases: 11 tumours (14.1%) were right sided, and 60 (76.9%) left sided. There was a significant difference in lesion location (p=0.001), with the MMR-deficient group representing more right-sided tumours.
T classification
In half of the tumours T stage could be identified. A T stage of more than T2 was identified in 91.7% of tumours with gene product deficiency and in 57.1% of tumours with gene product. There was a significant difference (p=0.019) in MMR status by T stage (>T2 and <T2), with a higher T stage in the gene product-deficient tumours.
Number of tumours
In 3 cases there were multiple lesions; 2 with one other lesion and 1 with two other lesions. In all 3 cases hMLH1 and hMSH2 were present.
Discussion
We chose IHC for this study because this method is an accurate marker for MMR gene product deficiency, is available in most pathology laboratories and is relatively cheap.[14] Mutations in hMLH1 and hMSH2 occur in 80 - 90% of all LS cases. We chose to detect mutations in these two genes to identify patients with inherited CRC (specifically LS).
Although all LS cancers are associated with mutations in the MMR genes, sporadic cancers can also follow an MMR (gene mutation) pathway. It is estimated that up to 15% of sporadic cancers will follow this pathway.[15,16,23-25] In around 80% of these MSI-high tumours with hMLH1 deficiency, DNA MMR genes are inactivated due to promoter hypermethylation of the hMLH1 gene.[25-27] Neither MSI nor IHC can differentiate between cancers caused by LS and sporadic cancers that follow the MMR pathway.
We found 21.8% of all CRCs in the Northern Cape to be deficient for hMLH1 or hMSH2. In 14.1% of tissue analysed, tumours were deficient for hMLH1, and in 7.7% hMSH2 was absent. One would expect that around 80% of the 11 hMLH1- deficient tumours would show protein loss due to hypermethylation of hMLH1 promoter (i.e. sporadic mutation), and one could therefore estimate that 10.5% of all CRCs in the Northern Cape would be expected to be MMR gene product-deficient due to germline mutations.
This estimated frequency of hMLH1 and hMSH2 deficiency is approximately two to four times the reported rate in high-incidence areas.[9,11,12] These results support our hypothesis that in a low-incidence area the inherited form contributes a greater proportion of the overall CRC burden as a result of a reduction in the incidence of sporadic CRC.
LS is characterised by several properties such as colonic and extra-colonic tumours at an early age of onset (<50 years); synchronous and metachronous CRC; mucinous and signet ring tumours; predominance of right-sided CRC; and poor differentiation.[10] In this study we used these characteristics, apart from the presence of hMLH1 and hMSH2, to identify MMR-deficient tumours. Unfortunately inadequate family history did not allow us to use the Bethesda or Amsterdam guidelines.
In the group of tumours with MMR deficiency, more patients were younger (<50 years) at time of diagnosis (41.2% v. 22.0%) although analysis did not show a statistically significant difference. A mean age of 56.9 years (SD 14 years) was determined (range 31 - 76 years) in the gene product-deficient tumours. This mean age was lower than the 58.9 years (SD 16 years) in the group with presence of gene product, but the difference was not significant. Hampel et al.[27] reported a mean age of 50.4 years. Mucinous and signet ring cells were more often identified (p=0.041) in the gene product-deficient group (23.5% v. 6.6%), and more tumours were poorly differentiated (35.7% v. 5.7%) (p=0.002). T stage was higher (p=0.019) in this group as well, and tumours were more often right-sided (35.3% v. 6.6%) (p=0.001). These results support our hypothesis that CRC tumours deficient for hMLH1 and hMSH2 are inherited.
Identifying LS families, and detecting the individuals who are mutation-positive, is necessary, as colonoscopic screening of those found to carry the predisposing mutation can enable early detection of CRC and reduce mortality by 65%.[28-29] These patients have a lifetime risk of developing colorectal and extra-colonic cancer associated with LS of over 90%,[30] and lifelong surveillance for these cancers is indicated.[9,10] The possible high frequency of inherited CRC in this study shows the importance of identification of potential carriers of germline mutations in all low-incidence areas.
A few comments should be made concerning our research. In this study we chose to test for the products of the hMLHl and hMSH2 genes only. These two genes account for almost 80 - 90% of all identified mutations in LS, meaning that this method does not identify mutations in all MMR genes responsible for LS and underestimates the total number of LS cases.[10]
It should also be noted that the group of hMLH1-deficient tumours in this study may include some sporadic tumours. As mentioned above, a number of MSI-high tumours that do not stain for the hMLHl product have inactivated MMR genes due to hMLHl promoter hypermethylation. This is why absence of hMLH1 with IHC on its own is not specific for LS.
A proportion of all tumours in this study showing hMLH1 deficiency are expected to be sporadic. The presence of hMLHl hypermethylation does not exclude the possibility of a germline mutation,[25,31] but determination of BRAF mutations can be used to detect LS, as the presence of a BRAF mutation argues against LS.[15,32]
In conclusion, our study provides data suggesting that the frequency of MMR deficiency in CRC is relatively high in an area with a low incidence of CRC compared with high-incidence areas in the Western world. These results mean that if a CRC surveillance programme is to be effective, it is important to identify LS patients in a low-incidence area.[29]
The proportion of MMR-deficient CRCs of 10.5% in this study is based on a calculation, and a future study needs to be done to confirm that these tumours with MMR deficiency have germline mutations. Testing for a BRAF mutation is necessary to eliminate the sporadic tumours in all hMLH1-deficient cases. On all tumours showing hMLH1 and hMSH2, gene product IHC testing for MMR proteins hMSH6 and hPMS2 needs to be performed to identify missed MMR gene-deficient tumours.
Acknowledgements. The authors thank L Daniels of the NHLS in Kimberley, A Visser of the NHLS in Cape Town, and M Tyler of the Department of Surgical Research, University of Cape Town. The Cancer Association of South Africa and the Medical Research
Council funded the original research leading to the identification of genetic defects in LS in South Africa.
References
1. Center MM, Jemal A, Smith RA, Ward E. Worldwide variations in colorectal cancer. CA Cancer J Clin 2009;59(6):366-378. [http://dx.doi.org/10.3322/caac.20038] [ Links ]
2. Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin 2011;61(2):69-90. [http://dx.doi.org/10.3322/caac.20107] [ Links ]
3. Mqoqi N, Kellett P, Sitas F, Jula M. Incidence of histologically diagnosed cancer in South Africa, 1998 - 1999. National Cancer Registry of South Africa, 2004. [ Links ]
4. Wentink MQ, Rakers M, Stupart DA, Algar U, Ramesar R, Goldberg PA. Incidence and histological features of colorectal cancer in the Northern Cape Province, South Africa. S Afr J Surg 2010;48(4):109-113. [ Links ]
5. Ferlay J, Autier P, Boniol M, Heanue M, Colombet M, Boyle P. Estimates of the cancer incidence and mortality in Europe in 2006. Ann Oncol 2007;18(3):581-592. [http://dx.doi.org/10.1093/annonc/mdl498] [ Links ]
6. Howlader N, Noone AM, Krapcho M, eds. SEER Cancer Statistics Review, 1975-2008. Bethesda, MD: National Cancer Institute. Based on November 2010 SEER data submission, posted to the SEER website, 2011. http://seer.cancer.gov/csr/1975_2008/ (accessed 6 February 2013). [ Links ]
7. Boyle P, Levin B. IARC World Cancer Report 2008. Lyon: International Agency for Research on Cancer, 2011. [ Links ]
8. Calvert PM, Frucht H. The genetics of colorectal cancer. Ann Intern Med 2002;137(7):603-612. [ Links ]
9. Lynch HT, Riley BD, Weissman SM, et al. Hereditary nonpolyposis colorectal carcinoma (HNPCC) and HNPCC-like families: Problems in diagnosis, surveillance, and management. Cancer 2004;100(1):53-64. [http://dx.doi.org/10.1002/cncr.11912]
10. Lynch HT, de la Chapelle A. Hereditary colorectal cancer. N Engl J Med 2003;348(10):919-932. [http://dx.doi.org/10.1056/NEJMra012242] [ Links ]
11. Half E, Bercovich D, Rozen P. Familial adenomatous polyposis. Orphanet J Rare Dis 2009;4:22. [http://dx.doi.org/10.1186/1750-1172-4-22] [ Links ]
12. Vasen HF, Moslein G, Alonso A, et al. Recommendations to improve identification of hereditary and familial colorectal cancer in Europe. Fam Cancer 2010;9(2):109-115. [http://dx.doi.org/10.1007/s10689-009-9291-3] [ Links ]
13. Jasperson KW, Tuohy TM, Neklason DW, Burt RW. Hereditary and familial colon cancer. Gastroenterology 2010;138(6):2044-2058. [http://dx.doi.org/10.1053/j.gastro.2010.01.054] [ Links ]
14. Shia J. Immunohistochemistry versus microsatellite instability testing for screening colorectal cancer patients at risk for hereditary nonpolyposis colorectal cancer syndrome. Part I. The utility of immunohistochemistry. J Mol Diagn 2008;10(4):293-300. [http://dx.doi.org/10.2353/jmoldx.2008.080031] [ Links ]
15. Zhang L. Immunohistochemistry versus microsatellite instability testing for screening colorectal cancer patients at risk for hereditary nonpolyposis colorectal cancer syndrome. Part II. The utility of microsatellite instability testing. J Mol Diagn 2008;10(4):301-307. [http://dx.doi.org/10.2353/jmoldx.2008.080062] [ Links ]
16. Boland CR, Goel A. Microsatellite instability in colorectal cancer. Gastroenterology 2010;138(6):2073-2087. [http://dx.doi.org/10.1053/j.gastro.2009.12.064] [ Links ]
17. Hampel H, Frankel WL, Martin E, et al. Feasibility of screening for Lynch syndrome among patients with colorectal cancer. J Clin Oncol 2008;26(35):5783-5788. [http://dx.doi.org/10.1200/JCO.2008.17.5950] [ Links ]
18. Culling CFA. Handbook of Histopathological and Histochemical Techniques. 3rd ed. London, Butterworths & Co, 1974. [ Links ]
19. Boland CR, Koi M, Chang DK, Carethers JM. The biochemical basis of microsatellite instability and abnormal immunohistochemistry and clinical behavior in Lynch syndrome: From bench to bedside. Fam Cancer 2008;7(1):41-52. [http://dx.doi.org/10.1007/s10689-007-9145-9Links ] Arial, Helvetica, sans-serif" size="2">]
20. Hsieh P, Yamane K. DNA mismatch repair: Molecular mechanism, cancer, and ageing. Mech Ageing Dev 2008;129(7-8):391-407. [http://dx.doi.org/10.1016/j.mad.2008.02.012]
21. Hameed F, Goldberg PA, Hall P, Algar U, van Wíjk R, Ramesar R. Immunohistochemistry detects mismatch repair gene defects in colorectal cancer. Colorectal Dis 2006;8(5):411-417. [http://dx.doi.org/10.1111/j.1463-1318.2006.00956.x] [ Links ]
22. Washington MK, Berlin J, Branton P, et al. Protocol for the examination of specimens from patients with primary carcinoma of the colon and rectum. ArchPathol Lab Med 2009;133(10):1539-1551. [ Links ]
23. Wright CM, Dent OF, Barker M, et al. Prognostic significance of extensive microsatellite instability in sporadic clinicopathological stage C colorectal cancer. BrJ Surg 2000;87(9):1197-1202. [http://dx.doi.org/10.1046/j.1365-2168.2000.01508.x] [ Links ]
24. Aaltonen LA, Salovaara R, Kristo P, et al. Incidence of hereditary nonpolyposis colorectal cancer and the feasibility of molecular screening for the disease. N Engl JMed 1998;338(21):1481-1487. [http://dx.doi.org/10.1056/NEJM199805213382101] [ Links ]
25. Herman JG, Umar A, Polyak K, et al. Incidence and functional consequences of hMLH1 promoter hypermethylation in colorectal carcinoma. Proc Natl Acad SciUSA 1998;95(12):6870-6875. [http://dx.doi.org/10.1073/pnas.95.12.6870] [ Links ]
26. Cunningham JM, Kim CY, Christensen ER, et al. The frequency of hereditary defective mismatch repair in a prospective series of unselected colorectal carcinomas. Am J Hum Genet 2001;69(4):780-790. [http://dx.doi.org/10.1086/323658] [ Links ]
27. Hampel H, Frankel WL, Martin E, et al. Screening for the Lynch syndrome (hereditary nonpolyposis colorectal cancer). N Engl J Med 2005;352(18):1851-1860. [http://dx.doi.org/10.1056/NEJMoa043146] [ Links ]
28. Jarvinen HJ, Aarnio M, Mustonen H, et al. Controlled 15-year trial on screening for colorectal cancer in families with hereditary nonpolyposis colorectal cancer.Gastroenterology 2000;118(5):829-834. [http://dx.doi.org/10.1016/S0016-5085(00)70168-5] [ Links ]
29. Stupart DA, Goldberg PA, Algar U, Ramesar R. Surveillance colonoscopy improves survival in a cohort of subjects with a single mismatch repair gene mutation. Colorectal Dis 2009;11(2):126-130. [http://dx.doi.org/10.1111/j.1463-1318.2008.01702.x] [ Links ]
30. Vasen HF, Wijnen JT, Menko FH, et al. Cancer risk in families with hereditary nonpolyposis colorectal cancer diagnosed by mutation analysis. Gastroenterology 1996;110(4):1020-1027. [http://dx.doi.org/10.1053/gast.1996.v110.pm8612988] [ Links ]
31. Rahner N, Friedrichs N, Steinke V, et al. Coexisting somatic promoter hypermethylation and pathogenic MLH1 germline mutation in Lynch syndrome. J Pathol 2008;214(1):10-16. [ Links ]
32. Nakagawa H, Nagasaka T, Cullings HM, et al. Efficient molecular screening of Lynch syndrome by specific 3' promoter methylation of the MLH1 or BRAF mutation in colorectal cancer with high-frequency microsatellite instability. Oncol Rep 2009;21(6):1577-1583. [ Links ]
 Corresponding author:
Corresponding author:
P A Goldberg
(paul.goldberg@uct.ac.za)


{kind=link}