










Servicios Personalizados
Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkSouth African Journal of Science
versión On-line ISSN 1996-7489
versión impresa ISSN 0038-2353
S. Afr. j. sci. vol.116 no.1-2 Pretoria ene./feb. 2020
http://dx.doi.org/10.17159/sajs.2020/6313
RESEARCH ARTICLE
Spatio-seasonal variations in the faecal bacterial community of Zulu sheep grazing in communally managed rangeland
Thembinkosi G. XuluI; Obinna T. EzeokoliII, III; Arvind K. GuptaII, *; Charlotte MienieII; Cornelius C. BezuidenhoutII; Nokuthula W. KuneneI
IDepartment of Agriculture, University of Zululand, KwaDlangezwa, South Africa
IIUnit for Environmental Sciences and Management, North-West University, Potchefstroom, South Africa
IIIMicrobiology and Environmental Biotechnology Research Group, Agricultural Research Council - Institute for Soil, Climate and Water, Pretoria, South Africa
ABSTRACT
The adaptation of Zulu (Nguni) sheep (Ovis aries) to environmental stress and survival under extensive conditions makes them uniquely important to rural Nguni farmers of South Africa. Here, the faecal bacterial community of five Zulu sheep populations managed under extensive conditions across summer and winter seasons was investigated in order to understand the influence of prevailing seasonal factors. Bacterial operational taxonomic units (OTUs)/species (at 97% 16S rRNA gene similarity) in Zulu sheep faeces were more diverse in winter than in summer at most (80%) sites and varied between seasons at specific sites. Firmicutes was the most abundant phyla in both summer and winter seasons, while the relative abundance of Actinobacteria reduced in 80% of sites from summer to winter. The genera (or family) such as Akkermansia, Eubacterium coprostanoligenes group, Intestinibacter, R-7 group (family Christensenellaceae), Ruminococcus, Ruminoclostridium, Treponema and UCG-005 (family Ruminococcaceae) were relatively more abundant and belonged to a 'core microbiome' of Zulu sheep faeces. Between seasons, Acinetobacter, Jeotgalicoccus, Methanobrevibacter, Phascolarctobacterium and Planomicrobium were differentially abundant. Overall, results suggest increased richness and diversity of bacteria from summer to winter which may be related to spatio-seasonal variations in grazing management, forage types and availability. This observation serves as baseline evidence, justifying further controlled studies investigating, amongst other factors, effects of forage type and availability across seasons on ruminal microbiota of Zulu sheep grazing in communally managed rangelands.
SIGNIFICANCE:
•Spatio-seasonal dynamics in the bacterial community of Zulu sheep faeces suggest differences in forage type and availability across sites potentially influence faecal bacteria of Zulu sheep.
•The study provides a basis for further controlled studies investigating the influence of environmental factors on rumen and faecal microbiomes of Zulu sheep
Keywords: 16S rRNA gene diversity, culture-independent analysis, ruminants, high-throughput sequencing
Introduction
Zulu sheep (Ovis aries) are one of the oldest and most prominent indigenous Nguni sheep in KwaZulu-Natal Province, South Africa. These sheep play a major role in the livelihood of rural farmers, including being a source of meat, manure, hides and income as well as serving socio-cultural purposes.1,2 The Zulu sheep breed possesses traits for survival and adaptation to environmental stresses such as drought and animal diseases peculiar to the KwaZulu-Natal region of South Africa.3 The sheep are commonly grazed under extensive production on marginal ecological areas which are not suitable for crop cultivation.1,4 Moreover, animal production under extensive agriculture is notably influenced by several environmental conditions, which include changes in forage availability and weather patterns (or seasons).5-9 This necessitates the seasonal assessment of the nutritional requirements and health status of animals in pastures.
Recent studies in ruminants indicate that the rumen microbiome varies with diet and host breed.10 According to McSweeney and Mackie11, ruminants and their gut-associated microbes have mutually co-evolved while adapting to climatic and botanic environments. The mutualistic contributions of rumen microbes include the breakdown of substrates (which the ruminant host cannot normally metabolise) and the synthesis of essential vitamins.12,13 Overall, rumen microbes contribute to the animal's well-being by performing nutritional, physiological, immunity and protective functions.11,12,14
Unfortunately, the composition and abundance of this functionally important gut microbial species can be altered by several factors, which include antibiotic use, age of the animal, geographical location, seasonal changes, feeding regimes, forage quality and the health of the host animal.15-17 Such factors may predispose increased faecal shedding of rumen microbiota, including some pathogenic species.18-20 For example, cold stress in animals5,6,9,17 could predispose the migration of microbial cells from the rumen to the lower tract6,21. Similarly, the faecal shedding of some bacterial species, including Escherichia coli and Listeria monocytogenes, has been correlated with an increase in temperature and antibiotic administration in dairy cows.19,22 Presently, there is little or no information on the faecal microbiota of indigenous Zulu sheep breeds grazing in their native environment. It is also unknown how seasonal factors influence microbial shedding in the faeces of Zulu sheep populations in communally managed pastures. Therefore, this pilot study aimed to investigate the spatial and seasonal variation in the faecal bacterial community of Zulu sheep grazing in pasture-based systems. This pilot study serves to present baseline data for further controlled studies investigating the effect of forage type and environmental conditions on the dynamics of the rumen and faecal bacteria of Zulu sheep.
Materials and methods
Study sites and sample collection
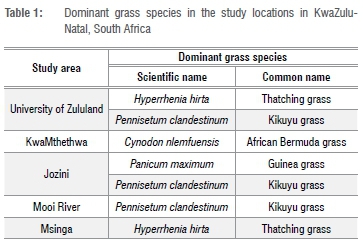
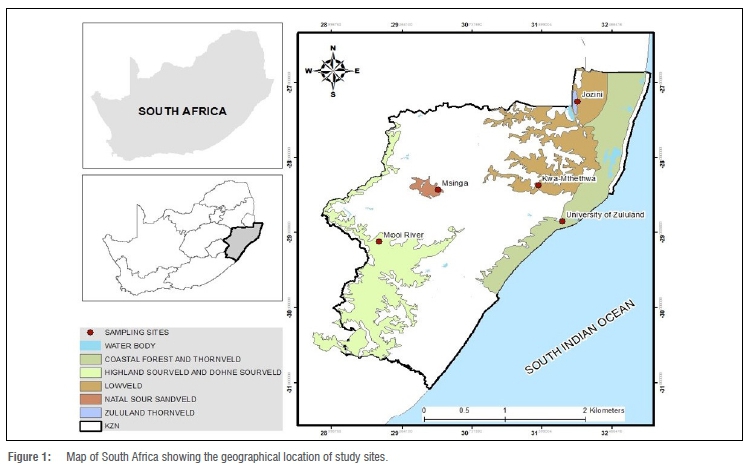
The Zulu sheep populations sampled were from five communally managed rangeland-based production systems (extensive conditions) in KwaZulu-Natal Province of South Africa: Mooi River (MR), Msinga (MSI), Jozini (JOZ), KwaMthethwa (MTH) and the University of Zululand (UZ) (Figure 1). The size of the herd at all locations ranged from an estimated 55 to 120 sheep. These study sites are located at altitudes of 90 m to 1900 m above sea level. Annual rainfall for this area ranges from 600 mm to about 1400 mm, while temperature ranges from 16 °C to 25 °C in winter (June to August) and from 23 °C to 33 °C in summer (mid-October to February).23 Winter months are cold and dry, while summer months are warm and wet. The forage grasses at these sites include Hyperrhenia hirta (thatching grass), Pennisetum clandestinum (Kikuyu grass), Cynodon nlemfuensis (African Bermuda grass) and Panicum maximum (Guinea grass) (Table 1).
In each of the five sites, 20 healthy (based on physical inspection) adult sheep were randomly sampled (without respect to sex) in the summer (October/November) and winter (June/July) seasons of 2014 and 2015, respectively, amounting to a total of 200 samples. For each individual sheep, faecal samples were collected aseptically from the rectum and immediately placed on ice. All procedures performed on animals during sample collection were in accordance with the ethical standards of the UniZulu Research Ethics Committee (certificate number UZREC171110-030 PGM 2015/250). Samples were stored at -20 °C in the laboratory prior to genomic DNA extraction.
Extraction of genomic DNA
Genomic DNA was extracted directly from approximately 150 mg of the faecal sample by using the ZR Fecal DNA MiniPrep Extraction kit (Zymo Research, Irvine, CA, USA) according to the instructions of the manufacturer. The integrity of DNA was verified by agarose gel electrophoresis while DNA concentration was determined using a NanoDrop Spectrophotometer (ND1000, NanoDrop Technologies Inc., Wilmington, DE, USA). DNA was stored at -20 °C prior to downstream analysis.
16S rRNA gene library preparation
The bacterial community in Zulu sheep faeces was analysed using high-throughput sequencing of the partial 16S rRNA gene (hypervariable region V3-V4) on the Illumina MiSeq sequencer (Illumina Inc., CA, USA). A partial 16S rRNA gene library was constructed by using universal primers 341F (forward) and 805R (reverse)24 as previously described25,26. Because an objective of this pilot study was to obtain an overview of the core bacterial population in each sampling location per season, all genomic DNA of samples from each location per season (sample size, n=20) were pooled in equal proportions (equimolar basis) prior to library preparation. Library preparation was then duplicated for each DNA pool. Following library preparation, a 2 x 300-bp paired-end sequencing run was performed on the Illumina MiSeq sequencer (Illumina Inc., CA, USA). The DNA extraction, library preparation and Illumina sequencing were performed at the Microbiology Group of the Unit for Environmental Sciences and Management, North-West University, Potchefstroom, South Africa.
Bioinformatic analyses
Sequence reads were first de-multiplexed and trimmed of primers and barcode sequences by using the on-board MiSeq reporter software (Illumina Inc., CA, USA). Sequence quality was further assessed using Fastqc (Babraham Bioinformatics, UK; https://www.bioinformatics.babraham.ac.uk/index.html) prior to assembling paired reads and quality-filtering to remove sequences with ambiguous bases and spurious length by using PANDAseq software.27 Thereafter, assembled quality-filtered reads were clustered into operational taxonomic units (OTUs) at 97% 16S rRNA gene sequence similarity by using the closed-reference OTU picking script in Quantitative Insight into Microbial Ecology (QIIME) software (version 1.9)28 and by aligning against the Silvangs rRNA database (release 123)29. Similarly, the taxonomic assignment of OTUs was performed using the Silvangs rRNA database taxonomy. The OTU table was depleted of singletons and rarefied to a single depth before computing alpha and beta diversities in QIIME software. Alpha diversity indices computed included observed OTUs (richness), Chao1 and Shannon-Wiener index of diversity (H'). For beta diversity, Bray-Curtis dissimilarity distances between samples was computed using the vegan package (version 2.5.5)30 of R software (version 3.5.3)31 and subsequently subjected to principal coordinate analyses by using the 'ape' package (version 5.3)32. Additionally, a core microbiome analysis was performed in QIIME to determine the bacterial phylotypes which are present and relatively dominant in all faecal samples.
Statistical analysis
Except otherwise stated, all statistical analyses were performed in R software. Alpha diversity indices were subjected to Wilcoxon rank-sum test for comparison between seasons and to Kruskal-Wallis H-test for comparisons across sites. Statistical tests for differences in bacterial community composition and structure amongst groups were performed by using a two-way (season x site) permutational multivariate analysis of variance (PERMANOVA) by using the 'adonis ()' function of the vegan package. Permutational test of homogeneity of multivariate dispersion (PERMDISP) was further used to test the difference in spread among groups. Before multivariate analysis, singletons and OTUs present in only one sample were eliminated by using the 'dropsec ()' function in the labdsv package (version 1.8.0).33 Multivariate analyses were performed on the log10-transformed relative proportion of each OTU within a sample by using the 'decostand ()' function in the labdsv package of R software. The log-transformation was of the order log10 (x) +1, where x > 0.34 Lastly, the linear discriminant analysis effect size (LEfSe)35 was used to determine differential abundant taxa between seasons as well as amongst sites. For LEfSe, default parameters (i.e. Wilcoxon rank-sum test or Kruskal-Wallis test p<0.05, linear discriminant analyses (LDA) >2.0) were used. The output from LEfSe was further visualised as an annotated cladogram using GraPhlAn.36
Data accessibility
Raw sequence reads obtained in this study are available in the Sequence Read Archive of the US National Center for Biotechnology Information (https://www.ncbi.nlm.nih.gov/sra) under the BioProject accession number PRJNA356736.
Henceforth, wherever applicable, abbreviated site names with 'W' appended at the end (e.g. MRW, MSIW, JOZW, MTW and UZW) denote winter samples, while abbreviated site names with 'S' appended at the end (e.g. MRS, MSIS, JOZS, MTS and UZS) denote summer samples. Duplicate samples have a '2' added after 'W' or 'S'.
Results
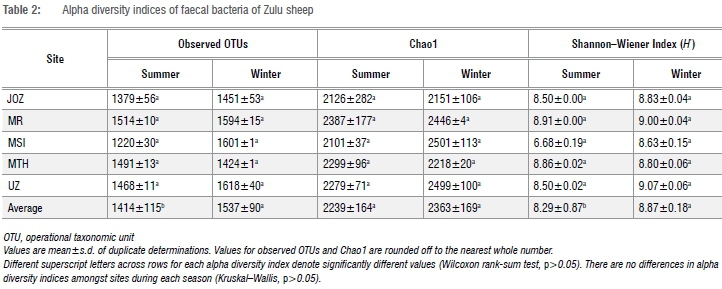
Spatio-seasonal comparison of alpha diversity OTUs in Zulu sheep faeces
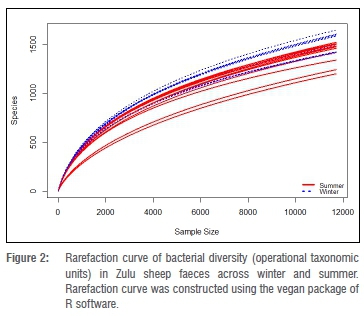
In total, 2 151 796 sequence reads were obtained after quality trimming and assignment of reads into OTUs. Following rarefaction (without replacement) of sequences at a depth of 11 700 sequences per sample, a total of 5530 OTUs (1385 OTUs unique to summer, 1379 OTUs unique to winter and 2766 shared OTUs) were obtained in all samples (data not shown). The richness, Chao1 richness estimation and Shannon-Wiener index of diversity for OTUs in faeces of each Zulu sheep population did not significantly differ between summer and winter (Wilcoxon rank-sum test, p>0.05) (Table 2). Similarly, differences in these alpha diversity measures were not significant (Kruskal-Wallis test, p>0.05) across sites during either summer or winter. However, higher OTU richness and diversity were observed in winter compared to summer in all but one (Msinga) location (Table 2). Overall, these results suggest that the faecal bacterial community of Zulu sheep is more diverse in winter, which may indicate increased shedding of bacteria in faeces during the winter season compared to the summer season. The non-significant (p>0.05) difference in Chao1 but significant differences in richness and Shannon-Wiener index of diversity between overall winter and summer faecal OTUs may be due to the lack of sufficient sub-sampling depth as revealed by the rarefaction curve of Figure 2. Thus, indications are that the observed richness and diversity of OTUs are underestimated.
The unweighted (absence/absence of taxa) and weighted (absence/absence and relative abundance of taxa) Bray-Curtis distance's principal coordinates analysis (PCoA) plots of the OTUs distributions (at 97% 16S rRNA sequence similarity) shown in Figure 3 indicate that the bacterial community in Zulu sheep faeces at given sites differed between winter and summer seasons. Distinctively, the bacterial community composition (unweighted) and structure (weighted) of faeces in summer were very dissimilar from their winter counterparts in Msinga and Jozini sites (Figure 3a,b), suggesting that large differences exist in the type and abundance of bacteria shed in the Zulu sheep faeces between winter and summer. However, the close associations in the faecal bacterial community of Zulu sheep in Mooi River rangelands during winter and summer may suggest that prevailing environmental conditions in this location are similar across winter and summer seasons.

PERMANOVA of both unweighted and weighted Bray-Curtis dissimilarities between bacterial OTU diversity of Zulu sheep faeces show that effects of interaction between factors (site and season) are significant (p<0.001) for both unweighted and weighted Bray-Curtis dissimilarities between sample groups. However, a significant PERMDISP (p<0.001) result observed for individual effects suggests that differences between sites and seasons may be due to lack of homogeneity in spread within each sample group. Thus, the 'location' and 'site' effects on bacterial community composition and structure suggested by PERMANOVA may be due to other co-founding or random variables, including sampling and site-specific variations. This assumption is further supported by a PERMANOVA r-square value which indicates that the site × season interaction accounts for only 24.4% and 29.0% variations in the Zulu sheep faecal bacterial community composition (unweighted) and structure (weighted), respectively.
Taxonomic diversity of relatively abundant and core phylotypes of Zulu sheep faecal bacteria
Most of the OTUs were taxonomically assigned to 21 phyla (Figure 4). In Figure 4, the relative abundance of phyla in Zulu sheep faeces differs among sites and between seasons. Firmicutes was the most abundant phylum of the faeces in summer (64.96±5.32%) and winter (64.52±9.36%) (Figure 4).
The phylum Bacteroidetes was the second most abundant phylum in winter at all sites, and in summer at all but the Msinga site, where Actinobacteria was the second most abundant phylum (Figure 4). Other phyla that constituted at least 1% relative abundance of the Zulu sheep faecal bacteria in both summer and winter included Proteobacteria, Spirochaetae and Verrucomicrobia. On average, the Firmicutes: Bacteroidetes ratio was approximately 4:1 (64.96±5.32%: 15.39±8.29%) in summer and 2:1 (64.52±9.36%: 26.02±8.74%) in winter. From summer to winter (the chronological order of sample collection), the Actinobacteria phylum population generally reduced in the faeces of Zulu sheep in a majority (80%) of the sites (Figure 4).
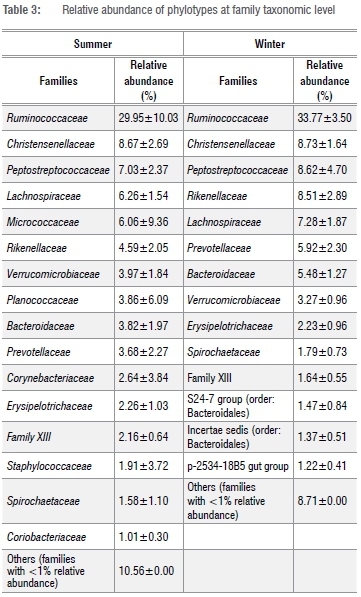
At the family taxa level, Ruminococcaceae was the most abundant in summer (29.95±10.03%) and winter (33.77±3.50%) (Table 3). Other families, which constituted at least 5% relative abundance of OTUs in either summer or winter included Bacteroideceae, Christensenellaceae, Lachinospiraceae, Micrococcaceae, Peptostreptococcaceae, Planococcaceae, Prevotellaceae and Rikenellaceae (Table 3). Across locations, Rumminococcaceae was the most abundant family in the faeces of Zulu sheep in the winter. However, in the summer, Rumminococcaceae was the most abundant in only four of the five study sites; in the remaining one location (Msinga), Planococcaceae was the most abundant family (data not shown).
The relative abundance of OTUs at the genera taxa level (family names are provided where OTUs are unclassified at genus taxa level) which constituted at least 1% maximum relative abundance in all study sites are shown in the heat map of Figure 5. Summarily, species of the genera (or family) such as R-7 group (family: Christensenellaceae), UCG-005 (family: Ruminococcaceae) and Intestinibacter were amongst the relatively more abundant genera across samples (Figure 5). In addition to the aforementioned relatively more abundant phylotypes, a core microbiome analysis (OTUs present in all samples) revealed that a majority of the phylotypes presented in Figure 5, including Akkermansia, Alistipes, Bacteroides, Eubacterium coprostanoligenes group, NK4A136 group (family: Lachnospiraceae), Phocaeicola, RC9 gut group (family: Rikenellaceae), Ruminoclostridium, Ruminococcus, UCG-003 (family: Prevotellaceae), UCG-004 (family: Prevotellaceae), UCG-010 (family: Ruminococcaceae), UCG-013 (family: Ruminococcaceae) and Treponema (Figure 5) constitute the 'core microbiome' of Zulu sheep faeces.

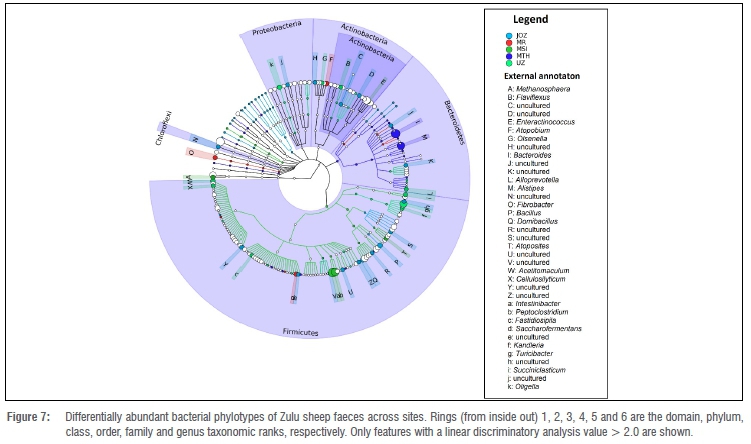
Spatio-seasonal differential abundance of faecal bacteria
The discriminant analyses revealed that a majority (75%) of the bacteria phylotypes which were discriminatory (Wilcoxon rank-sum test, p<0.05, LDA>2.0) between seasonal faecal bacterial communities of Zulu sheep were uncultured (Figure 6). The phylotypes (classifiable at the genus taxonomic rank) Acinetobacter, Jeotgalicoccus, Methanobrevibacter and Planomicrobium were differentially more abundant in summer than in winter (Figure 6), whereas Phascolarctobacterium was differentially more abundant in winter than summer (Figure 6). Across sites, a total of 99 features (or phylotypes) were discriminative (Kruskal-Wallis, p<0.05, LDA>2.0) across all taxonomic ranks (Figure 7). These phylotypes potentially drive the differences observed among the faecal bacterial community of Zulu sheep populations in multivariate space (Figure 3). In particular, Bacillus and Domibacillus were differentially most abundant in Jozina while Atopobium, Fibrobacter and Saccharofermentans were differentially most abundant in Mooi River. Similarly, Alloprevotella, Atopostipes, Intestinibacter, Methanosphaera, Olsenella, Peptoclostridium and Succiniclastium were differentially most abundant in Msinga, Alistipes and Bacteroides in KwaMthethwa, while Acetitomaculum, Cellulosilyticum, Enteractinococcus, Fastidiosipila, Flaviflexus, Kandleria, Oligella and Turicibacter were differentially most abundant in the faeces of the Zulu sheep population located at University of Zululand (Figure 7).
Discussion
The dynamics of the faecal microbiota community diversity of ruminants may be driven by several factors including variations in feeding operation, geographical availability and type of forage.18,19,22 In this study, high-throughput sequencing of the bacterial 16S rRNA gene was used to elucidate the faecal bacterial community dynamics of Zulu sheep populations during winter and summer seasons. Furthermore, the study provided an insight into the potential of Zulu sheep faeces to serve as a source of potential pathogens around grazing environments.
The observed higher faecal bacterial OTU richness and diversity in the winter compared to the summer in a majority (except KwaMthethwa) of the study sites suggest increased faecal shedding of Zulu sheep gut microbes during the winter season. Some studies have shown that cold stress may predispose the increased shedding of rumen bacteria.21,37,38 The observed higher OTU richness and diversity in winter may be predisposed by variation in diet across summer and winter. A previous study by Shanks et al.18 on the faecal microbiome of cattle fed in feedlots with different feeds - forage, processed grain and unprocessed grains - suggested that feeding operations or feed type were predictors of the faecal microbiome of cattle compared to the geographical location of the feedlot. Similarly, Callaway et al.39 reported that the inclusion of dried distiller grains in cattle feeds reduced faecal bacterial diversity in comparison to that in cattle which were fed a basal feedlot diet. These studies essentially show that a link exists between diet and the faecal microbiome of ruminants.
In rangelands such as in the current study, forage type and availability vary along seasonal lines. Between summer and winter, Zulu sheep feed on a variety of feed types, which subsequently predisposes variations in the population numbers of bacterial flora associated with the gut and faeces.39,40 For example, feed types have been suggested to increase the levels of certain bacteria in the faeces of cattle.40 Indeed, in the summer season, these regions are usually characterised by adequate agronomic conditions, which favour the growth of forage (grass) species on which the animals graze. It has been reported that the availability of grass species during the summer months influences the animals' preference for forage types. Such preferences include grazing on grass species in the summer to grazing on dry grasses and browsing on trees and shrubs in the dry winter months.41,42 However, the observed non-significant differences in the bacterial richness and diversity in faeces of most Zulu sheep populations between winter and summer suggest that any potential differences in feed type and availability between seasons are not determinants of the faecal bacterial richness and diversity of Zulu sheep. Nevertheless, the non-significant differences may also be due to the small sample number, pooling of samples in the present pilot study as well as the limitations of next-generation sequencing technology employed in the present study (discussed later).
In multivariate space, differences in bacterial community composition and structure were significantly influenced by an interaction amongst sites and between seasons. Possibly, the differences observed in the faecal bacterial community composition and structure across seasons, particularly in the Msinga Zulu sheep population may be due to the influence of seasonal variation in feed type and availability. Elsewhere, faecal microbial community composition of adult beef cattle was more shaped by feeding operations than by geographical location.18 However, Durso et al.43 observed a variation in the faecal bacterial communities of beef cattle which could not be linked to the influence of diet or weather. Further large-scale studies are required to validate the observations made in this study as well as to confirm factors which predispose the large variation (in multivariate space) between the summer and winter faecal bacterial community composition and structure observed in Msinga.18 In addition, because this study involved sheep breeds under extensive animal management, several co-founding variables - including environmental parameters, management practices, forage types and availability - render explanations for the faecal bacterial community diversity dynamics inconclusive. Further studies are required to determine the specific contribution and extent to which each of these factors influence the faecal bacterial community dynamics of Zulu sheep. Such further studies will assume a similar approach to that of recent investigations on the faecal microbiome of some ruminants.44,45
Firmicutes and Bacteroidetes phyla dominated the bacterial composition of Zulu sheep in both summer and winter seasons. Similar observations have been reported in previous microbiological studies of the faeces of ruminants, including beef and dairy cattle18,39,45,46 as well as sheep and goats44,47. In contrast, the high relative abundance of Actinobacteria in the faecal microbiome of Zulu sheep in the Msinga location is a rather surprising observation when compared to other sites (or populations), to its (Msinga population) winter faecal microbiome, and to other published studies.18,39,43,44,46,47 The disparity may suggest a stack contrast in prevailing conditions between winter and summer in Msinga, and from those of other sites. The relatively high composition of OTUs belonging to Actinobacteria phyla most likely account for the large bacterial community variation observed in multivariate space between summer samples from Msinga and those of its winter samples as well as those from other sites (Figure 3).
At the family taxa level, the majority of the OTUs belonged to the family Ruminococcaceae. The prevalence of bacteria belonging to the family Ruminococcaceae is not surprising considering the vast literature reporting their presence in ruminant stomach chambers48-50 as well as in faeces18,43,50. The abundance of Ruminococcaceae in ruminant faeces has been linked to carbohydrate-rich diets.10,51 It has been suggested that Ruminococcaceae, along with other families in the Zulu sheep faeces such as Lachinospiraceae and Prevotellaceae, predominantly contribute to ruminal biohydrogenation and breakdown of complex polysaccharides through the secretion of cellulolytic and hemicellulolytic enzymes.52-54 At the genus level, several genera, including Ruminococcus, Ruminoclostridium, Treponema, Akkermansia and Eubacterium coprostanoligenes were relatively abundant (≥1% maximum relative abundance) and present in all Zulu sheep faeces analysed. The presence of these species in all Zulu sheep faeces suggests that they constitute a 'core microbiome' of Zulu sheep faeces. Likewise, the presence of a core microbiome has been reported in previous faecal microbiome studies.18,44 These core genera largely belong to the families highlighted earlier and are therefore involved in the degradation of a wide range of organic matter, including forage feeds and complex polysaccharides.10,12,51
Season-wide and site-wide differential abundance analyses were performed to identify species which statistically varied in relative abundance between summer and winter as well as between sites. Such differential species are likely sensitive to the prevailing seasonal or site effect on Zulu sheep metabolism. For example, studies have shown that faecal shedding of pathogenic microbes may be correlated with temperature.19,22 Orpin et al.48 reported that the differences in forage type, feeding duration and rate as well as the flow rate of digesta through the rumen between summer and winter seasons may influence the ruminal microflora in Svalbard reindeer (Rangifer tarandus platyrhynchus). Deductively, such differences may also occur in summer and winter faeces of ruminants. More importantly, the differential shedding of these species may reflect conditions in the rumen39 which could have implications for animal production, particularly in terms of forage digestibility, feed nutrient utilisation and animal performance.13,55
More importantly, Zulu sheep faeces may serve as a source of pathogenic species in the vicinity in which they graze. In the present study, some abundant and differentially abundant phylotypes across season or site included genera of which some species are known human pathogens. Such genera include Enterococcus, Treponema and Peptoclostridium. Further studies utilising quantitative polymerase chain reaction (qPCR) of specific gene loci are required to detect and quantify the levels of potentially pathogenic species that may be present in Zulu sheep faeces. In addition, the isolation and characterisation of some of the differentially abundant bacteria may provide insights into their function and potential utilisation as bioindicators of ruminant health as well as for evaluating potential risks associated with Zulu sheep grazing on the environment.
The unavailability of detailed information on the prevailing management practices as well as feed type and quantities available to the Zulu sheep populations during the seasons constitute some of the limitations of this study. Such information is important in order to aid the understanding of possible factors driving the faecal bacteria dynamics across sites and season. As no approach is without limitations, the varying copy numbers of the 16S rRNA gene marker, the low-resolution of the next-generation sequencing approach to detect metabolically active but less dominant species, PCR bias, the inability to identify bacteria to species levels and the possibility of the sequence count rarefaction step to distort the true ecological diversity of a given environment56,57 are additional limitations of the study. Indeed, these limitations may influence the abundance estimates and richness of bacterial phylotypes reported. Nevertheless, the approach (next-generation sequencing of the 16S rRNA gene) is widely considered to be a robust and rapid method for profiling the bacterial community of any environment at a high-throughput scale.
In conclusion, the faecal bacterial community of Zulu sheep varied along spatio-seasonal lines, thereby suggesting differences in animal management practices and feed type, amongst other factors, across seasons and between locations. The faecal bacterial dynamics of specific species between seasons point towards similar dynamics in the bacterial communities along the stomach chambers of Zulu sheep. Further large-scale and controlled studies are required to investigate the effect of seasonal factors, including forage type and availability, as well as other management practices on the rumen bacterial community of Zulu sheep grazing in communally managed rangelands. Such studies will be important for maximising animal production through improving the digestibility of forage in the sheep rumen across seasonal lines.
Acknowledgements
This work was supported by the National Research Foundation (NRF) of South Africa (grant UID 76352), the University of Zululand and North-West University. We acknowledge an NRF bursary awarded to Thembinkosi Xulu (grant UID 105147) and postdoctoral research fellowship awarded by the North-West University to Arvind Gupta. We thank the rural Nguni farmers and the Department of Agriculture for their cooperation during sample collection, and Abram Mahlatsi and Lee Julies for technical assistance.
Authors' contributions
N.W.K. and C.C.B. conceived and supervised the study. T.G.X. conducted the sampling. T.G.X., A.K.G. and C.M. performed the laboratory analyses. O.T.E. performed the bioinformatics and statistical analyses. O.T.E. and T.G.X. wrote the first draft of the manuscript. All authors reviewed and approved the final manuscript.
References
1.Kunene N, Bezuidenhout C, Nsahlai I. Genetic and phenotypic diversity in Zulu sheep populations: Implications for exploitation and conservation. Small Rumin Res. 2009;84:100-107. https://doi.org/10.1016/j.smallrumres.2009.06.012 [ Links ]
2.Mavule B, Muchenje V, Kunene N. Characterization of Zulu sheep production system: Implications for conservation and improvement. Sci Res Essays. 2013;8:1226-1238. https://doi.org/10.5897/SRE2013.1872 [ Links ]
3.Kunene NW, Bezuidenhout CC, Nsahlai IV, Nesamvuni EA. A review of some characteristics, socio-economic aspects and utilization of Zulu sheep: Implications for conservation. Trop Anim Health Prod. 2011;43:1075-1079. https://doi.org/10.1007/s11250-011-9823-3 [ Links ]
4.Ramsay K, Harris L, Kotze A. Landrace breeds: South Africa's indigenous and locally developed farm animals. Pretoria: Farm Animal Conservation Trust; 2000. p. 38-39. [ Links ]
5.Ball DM, Collins M, Lacefield GD, Martin NP, Mertens DA, Olson KE, et al. Understanding forage quality. American Farm Bureau Federation Publication 1-01. Park Ridge, IL: American Farm Bureau Federation Publication; 2001. [ Links ]
6.Leng RA. Application of biotechnology to nutrition of animals in developing countries. FAO Animal production and health paper 90; Rome: FAO; 1991. [ Links ]
7.Nardone A, Ronchi B, Lacetera N, Ranieri MS, Bernabucci U. Effects of climate changes on animal production and sustainability of livestock systems. Livest Sci. 2010;130:57-69. https://doi.org/10.1016/j.livsci.2010.02.011 [ Links ]
8.Nyamukanza C, Scogings P, Kunene N. Forage-cattle relationships in a communally managed semi-arid savanna in northern Zululand, South Africa. Afr J Range Forage Sci. 2008;25:131-140. https://doi.org/10.2989/AJRF.2008.25.3.5.602 [ Links ]
9.Oelberg K. Factors affecting the nutritive value of range forage. J Range Manage. 1956;9:220-225. https://doi.org/10.2307/3894056 [ Links ]
10.Henderson G, Cox F, Ganesh S, Jonker A, Young W, Global Rumen Census Collaborators, et al. Rumen microbial community composition varies with diet and host, but a core microbiome is found across a wide geographical range. Sci Rep. 2015;5:14567. https://doi.org/10.1038/srep14567 [ Links ]
11.McSweeney C, Mackie R. Micro-organisms and ruminant digestion: State of knowledge, trends and future prospects. Background study paper no. FAO Commission on Genetic Resources for Food and Agriculture [document on the Internet]. c2012 [cited 2019 May 27]. Available from: http://www.fao.org/3/me992e/me992e.pdf [ Links ]
12.Malmuthuge N, Griebel PJ. The gut microbiome and its potential role in the development and function of newborn calf gastrointestinal tract. Front Vet Sci. 2015;2, Art. #36, 10 pages. https://doi.org/10.3389/fvets.2015.00036 [ Links ]
13.Wadhwa M, Bakshi M, Makkar HP. Modifying gut microbiomes in large ruminants: Opportunities in non-intensive husbandry systems. Anim Front. 2016;6(2):27-36. https://doi.org/10.2527/af.2016-0020 [ Links ]
14.Thirumalesh T, Krishnamoorthy U. Rumen microbial biomass synthesis and its importance in ruminant production. Int J Livest Res. 2013;3:5-26. https://doi.org/10.5455/ijlr.20130502081346 [ Links ]
15.Russell JB, Rychlik JL. Factors that alter rumen microbial ecology. Science. 2001;292:1119-1122. https://doi.org/10.1126/science.1058830 [ Links ]
16.Shakira G, Mirza I, Latif A. Scope of common DNA based methods for the study of rumen bacterial population. Bang J Anim Sci. 2013;41(2):141-146. https://doi.org/10.3329/bjas.v41i2.14134 [ Links ]
17.Waterman R, Grings E, Geary T, Roberts A, Alexander L, MacNeil M. Influence of seasonal forage quality on glucose kinetics of young beef cows. J Anim Sci. 2007;85(10):2582-2595. https://doi.org/10.2527/jas.2007-0023 [ Links ]
18.Shanks OC, Kelty CA, Archibeque S, Jenkins M, Newton RJ, McLellan SL, et al. Community structures of fecal bacteria in cattle from different animal feeding operations. Appl Environ Microbiol. 2011;77(9):2992-3001. https://doi.org/10.1128/AEM.02988-10 [ Links ]
19.Stenkamp-Strahm C, McConnel C, Rao S, Magnuson R, Hyatt D, Linke L. Climate, lactation, and treatment factors influence faecal shedding of Escherichia coli O157 pathotypes in dairy cows. Epidemiol Infect. 2017;145(1):115-125. https://doi.org/10.1017/S0950268816001928 [ Links ]
20.Wang O, McAllister TA, Plastow G, Stanford K, Selinger B. Interactions of the hindgut mucosa-associated microbiome with its host regulate shedding of Escherichia coli O157: H7 by cattle. Appl Environ Microbiol. 2018;84(1), e01738-17, 15 pages. https://doi.org/10.1128/AEM.01738-17 [ Links ]
21.Kennedy PM, Christopherson RJ, Milligan LP. Digestive responses to cold. In: Milligan LP, Grovum WL, Dobson A, editors. Control of digestion and metabolism in ruminants. Upper Saddle River, NJ: Prentice Hall; 1986. p. 285-306. [ Links ]
22.Bandelj P, Jamnikar‐Ciglenecki U, Ocepek M, Blagus R, Vengust M. Risk factors associated with fecal shedding of Listeria monocytogenes by dairy cows and calves. J Vet Intern Med. 2018;32(5):1773-1779. https://doi.org/10.1111/jvim.15234 [ Links ]
23.World Weather Online. Durban, KwaZulu-Natal monthly climate average, South Africa [webpage on the Internet]. No date [cited 2016 Dec 12]. Available from: https://www.worldweatheronline.com/durban-weather-averages/kwazulu-natal/za.aspx [ Links ]
24.Klindworth A, Pruesse E, Schweer T, Peplies J, Quast C, Horn M, et al. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013;41(1), e1, 11 pages. https://doi.org/10.1093/nar/gks808 [ Links ]
25.Mashiane RA, Ezeokoli OT, Adeleke RA, Bezuidenhout CC. Metagenomic analyses of bacterial endophytes associated with the phyllosphere of a Bt maize cultivar and its isogenic parental line from South Africa. World J Microbiol Biotechnol. 2017;33(4):80. https://doi.org/10.1007/s11274-017-2249-y [ Links ]
26.Ezekiel CN, Ayeni KI, Ezeokoli OT, Sulyok M, Van Wyk DA, Oyedele OA, et al. High-throughput sequence analyses of bacterial communities and multi-mycotoxin profiling during processing of different formulations of Kunu, a traditional fermented beverage. Front Microbiol. 2018;9, Art. #3282, 17 pages. https://doi.org/10.3389/fmicb.2018.03282 [ Links ]
27.Masella AP, Bartram AK, Truszkowski JM, Brown DG, Neufeld JD. PANDAseq: Paired-end assembler for illumina sequences. BMC Bioinform. 2012;13(1), Art. 31, 7 pages. https://doi.org/10.1186/1471-2105-13-31 [ Links ]
28.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7(5):335-336. https://doi.org/10.1038/nmeth.f.303 [ Links ]
29.Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, et al. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013;41:590-596. https://doi.org/10.1093/nar/gks1219 [ Links ]
30.Oksanen J, Blanchet FG, Kindt R, Legendre P, Minchin PR, O'Hara R, et al. Vegan: Community Ecology Package [software on the Internet]. No date [cited 2019 Oct 08]. Available from: https://CRAN.R-project.org/package=vegan [ Links ]
31.R Core Team. R: A language and environment for statistical computing. Vienna: R Foundation for Statistical Computing; 2017. [ Links ]
32.Paradis E, Claude J, Strimmer K. APE: Analyses of phylogenetics and evolution in R language. Bioinformatics. 2004;20(2):289-290. https://doi.org/10.1093/bioinformatics/btg412 [ Links ]
33.Roberts DW. labdsv: Ordination and multivariate analysis for ecology. Version 2016 [software on the Internet]. Available from: http://ecology.msu.montana.edu/labdsv/R [ Links ]
34.Anderson MJ, Ellingsen KE, McArdle BH. Multivariate dispersion as a measure of beta diversity. Ecol Lett. 2006;9:683-693. https://doi.org/10.1111/j.1461-0248.2006.00926.x [ Links ]
35.Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, et al. Metagenomic biomarker discovery and explanation. Genome Biol. 2011;12(6):R60. https://doi.org/10.1186/gb-2011-12-6-r60. [ Links ]
36.Asnicar F, Weingart G, Tickle TL, Huttenhower C, Segata N. Compact graphical representation of phylogenetic data and metadata with GraPhlAn. PeerJ. 2015;3, e1029, 17 pages. https://doi.org/10.7717/peerj.1029 [ Links ]
37.Nightingale KK, Fortes ED, Ho AJ, Schukken YH, Grohn YT, Wiedmann M. Evaluation of farm management practices as risk factors for clinical listeriosis and fecal shedding of Listeria monocytogenes in ruminants. J Am Vet Med Assoc. 2005;227(11):1808-1814. https://doi.org/10.2460/javma.2005.227.1808 [ Links ]
38.Romero-Pérez GA, Ominski KH, McAllister TA, Krause DO. Effect of environmental factors and influence of rumen and hindgut biogeography on bacterial communities in steers. Appl Environ Microbiol. 2011;77(1):258-268. https://doi.org/10.1128/AEM.01289-09 [ Links ]
39.Callaway T, Dowd S, Edrington T, Anderson R, Krueger N, Bauer N, et al. Evaluation of bacterial diversity in the rumen and feces of cattle fed different levels of dried distillers grains plus solubles using bacterial tag-encoded FLX amplicon pyrosequencing. J Anim Sci. 2010;88(12):3977-3983. https://doi.org/10.2527/jas.2010-2900 [ Links ]
40.Jacob M, Fox JT, Drouillard JS, Renter DG, Nagaraja TG. Effects of dried distillers' grain on fecal prevalence and growth of Escherichia coli O157 in batch culture fermentations from cattle. Appl Environ Microbiol. 2008;74(1):38-43. https://doi.org/10.1128/AEM.01842-07 [ Links ]
41.Bayer W, Alcock R, Dladla F, Gilles P, Masondo M, Mkhize P, et al. A study of indigenous livestock management in rural KwaZulu-Natal, South Africa. Mdukatshani: Mdukatshani Rural Development Project. Unpublished report 2004. [ Links ]
42.Peris K. Forage diversity and impact of grazing management on rangeland ecosystems in Mbeere district. Kenya Land Use Change Impacts and Dynamics (LUCID) Project Working Paper 36 [document on the Internet]. c2004 [cited 2019 Oct 07]. Available from: http://www.lucideastafrica.org/publications/Kamau_LUCID_WP36.pdf [ Links ]
43.Durso LM, Harhay GP, Smith TP, Bono JL, DeSantis TZ, Harhay DM, et al. Animal-to-animal variation in fecal microbial diversity among beef cattle. Appl Environ Microbiol. 2010;76(14):4858-4862. https://doi.org/10.1128/AEM.00207-10 [ Links ]
44.Donnell MMO, Harris HMB, Ross RP, O'Toole PW. Core fecal microbiota of domesticated herbivorous ruminant, hindgut fermenters, and monogastric animals. MicrobiologyOpen. 2017;6(5), e00509, 11 pages. https://doi.org/10.1002/mbo3.509 [ Links ]
45.Meale SJ, Li S, Azevedo P, Derakhshani H, Plaizier JC, Khafipour E, et al. Development of ruminal and fecal microbiomes are affected by weaning but not weaning strategy in dairy calves. Front Microbiol. 2016;7, Art. #582, 16 pages. https://doi.org/10.3389/fmicb.2016.00582 [ Links ]
46.Muñoz-Vargas L, Opiyo SO, Digianantonio R, Williams ML, Wijeratne A, Habing G. Fecal microbiome of periparturient dairy cattle and associations with the onset of Salmonella shedding. PLoS ONE. 2018;13(5), e0196171, 16 pages. https://doi.org/10.1371/journal.pone.0196171 [ Links ]
47.Tanca A, Fraumene C, Manghina V, Palomba A, Abbondio M, Deligios M, et al. Diversity and functions of the sheep faecal microbiota: A multi‐omic characterization. Microb Biotechnol. 2017;10(3):541-554. https://doi.org/10.1111/1751-7915.12462 [ Links ]
48.Orpin CG, Mathiesen SD, Greenwood Y, Blix AS. Seasonal changes in the ruminal microflora of the high-arctic Svalbard reindeer (Rangifer tarandus platyrhynchus). Appl Environ Microbiol. 1985;50(1):144-151. [ Links ]
49.Mathiesen SD, Orpin CG, Greenwood Y, Blix AS. Seasonal changes in the cecal microflora of the high-arctic Svalbard reindeer (Rangifer tarandus platyrhynchus). Appl Environ Microbiol. 1987;53(1):114-118. [ Links ]
50.Azad E, Derakhshani H, Forster R, Gruninger R, Acharya S, McAllister T, et al. Characterization of the rumen and fecal microbiome in bloated and non-bloated cattle grazing alfalfa pastures and subjected to bloat prevention strategies. Sci Rep. 2019;9(1), Art. #4272, 13 pages. https://doi.org/10.1038/s41598-019-41017-3 [ Links ]
51.Khafipour E, Li S, Tun H, Derakhshani H, Moossavi S, Plaizier J. Effects of grain feeding on microbiota in the digestive tract of cattle. Anim Front. 2016;6(2):13-19. https://doi.org/10.2527/af.2016-0018 [ Links ]
52.Flint HJ, Bayer EA, Rincon MT, Lamed R, White BA. Polysaccharide utilization by gut bacteria: Potential for new insights from genomic analysis. Nat Rev Microbiol. 2008;6(2):121-131. [ Links ]
53.Huws SA, Kim EJ, Lee MR, Scott MB, Tweed JK, Pinloche E, et al. As yet uncultured bacteria phylogenetically classified as Prevotella, Lachnospiraceae incertae sedis and unclassified Bacteroidales, Clostridiales and Ruminococcaceae may play a predominant role in ruminal biohydrogenation. Environ Microbiol. 2011;13(6):1500-1512. https://doi.org/10.1111/j.1462-2920.2011.02452.x [ Links ]
54.Biddle A, Stewart L, Blanchard J, Leschine S. Untangling the genetic basis of fibrolytic specialization by Lachnospiraceae and Ruminococcaceae in diverse gut communities. Diversity. 2013;5(3):627-640. https://doi.org/10.3390/d5030627 [ Links ]
55.Naya DE, Karasov WH. Food digestibility by microbes in wild ruminants: The effect of host species and dietary substrate. Rangelands. 2011;33(1):31-34. https://doi.org/10.2458/azu_rangelands_v33i1_naya [ Links ]
56.McMurdie PJ, Holmes S. Waste not, want not: Why rarefying microbiome data is inadmissible. PLoS Comput Biol. 2014;10(4), e1003531, 12 pages. https://doi.org/10.1371/journal.pcbi.1003531 [ Links ]
57.Ezeokoli OT, Adeleke R, Bezuidenhout CC. Core bacterial community of soy-daddawa: Insights from high-throughput DNA metabarcoding. LWT-Food Sci Technol. 2018;97:61-66. https://doi.org/10.1016/j.lwt.2018.06.039 [ Links ]
 Correspondence:
Correspondence:
Thembinkosi Xulu
mnqobixulu1@gmail.com
Received: 06 Aug. 2019
Revised: 11 Oct. 2019
Accepted: 18 Oct. 2019
Published: 29 Jan. 2020
Editors: Teresa Coutinho; Salmina Mokgehle
Funding: National Research Foundation (South Africa), University of Zululand, North-West University
* Current affiliation: Department of Biotechnology and Microbiology, AKS University, Satna, India
Supplementary Material
The open data set is available here: [Open data set]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}