










Servicios Personalizados
Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkSouth African Journal of Science
versión On-line ISSN 1996-7489
versión impresa ISSN 0038-2353
S. Afr. j. sci. vol.115 no.9-10 Pretoria sep./oct. 2019
http://dx.doi.org/10.17159/sajs.2019/5666
RESEARCH ARTICLE
Theoretical evaluation of valeraldehyde
Muhammad AzizI, II; Muhammad AnwarII; Shazia IqbalIII
ISchool of science, University of Management and Technology Lahore, Lahore, Pakistan
IIInstitute of Biochemistry, University of Balochistan, Quetta, Pakistan
IIIDepartment of Chemistry, Balochistan University of Information Technology, Engineering and Management Sciences, Quetta, Pakistan
ABSTRACT
The aim of this study was to compute the theoretical (software-based) spectroscopic properties of valeraldehyde as a member of the aldehyde family. The structural, thermochemical, electrical and spectroscopic properties of valeraldehyde were investigated using a quantum calculation approach. The infrared, ultraviolet-visible, Raman, and vibrational self-consistent field calculations and thermochemistry were calculated using the computational software GAMESS (General Atomic and Molecular Electronic Structure System) and the nuclear magnetic resonance predictions were calculated. The calculated energy gap between the lowest unoccupied molecular orbital (LUMO) and the highest occupied molecular orbital (HOMO) was 164.892.eV, which means valeraldehyde is a poor electrical conductor. The band gap is a non-neglected parameter for optical material. Results of the current computational analysis are useful to predict even a complex aldehyde precursor.
SIGNIFICANCE:
•The computational strategy used successfully here could also be useful for investigation of other aldehyde polymers
Keywords: DFT spectroscopy calculation, pentanal, aldehyde
Introduction
Valeraldehyde is a colourless member of the aldehyde family. Applications involving aldehydes are vast. Valeraldehyde holds prime importance in the food industry as a flavour enhancer and in the polymer industry.1 Vegetable glycerin produces aldehydes after heat contact in electronic cigarettes.2 Darker beers produce disproportionately high levels of Strecker aldehydes.3 However, the European Food Agency has prohibited the addition of flavouring agents4, including valeraldehyde. Although aldehydes occur naturally in cells and are generated during intermediary metabolism of natural compounds, metabolism of drugs and xenobiotics may result in toxic interactions, which is why the pharmaceutical industry has a keen interest in aldehyde studies.5
The aldehyde under consideration here is valeraldehyde. Valeraldehyde is neither a simple nor a complex member of the aldehyde family. With the introduction of each ethyl (C2H5), the molecule can alter its intrinsic properties to some extent, so more data are needed to provide guidance to scientists working with this molecule. Kinetic modelling studies have been devoted to understanding the oxidation chemistry of aldehydes, because of their importance as intermediate and product species in alkane and biofuel oxidation.6
Through the use of computer-aided material design, the end product and the choice of material from which the product is made are interdependent activities. In this way, complex products can be created easily and directly from simulating the base result, with minimal requirements for the design or manufacturing tool.7,8 Computational spectroscopic study is an analytical tool for identifying organic compounds. It is hoped that experimental methods in combination with computational methods will be a powerful tool for determining the absolute configuration of molecules. Through the use of computational software, vibrational spectroscopic simulations have long assisted in solving chemical problems and the industrial use of vibrational spectroscopy is highlighted by many.9 Vibrational spectroscopy is based on the methods of theoretical chemistry consolidated into effective computer programs to determine the structural properties of a compound. Advancements in computational analytical chemistry base software has paved the way to predicting results before undertaking experimental analysis, which creates greater confidence in the results of the analysis.10 Different types of computational methods and models are available. Selecting the appropriate approach for a particular molecule is the task of the computational chemist. Density functional theory is used to determine electronic properties with reference to β-functionalisation of aldehydes, as reported by Liu et al.11 To our knowledge, this is the first report of infrared (IR), ultraviolet-visible (UV-Vis), Raman and vibrational self-consistent field (VSCF) spectrograms of valeraldehyde using density functional theory. The International Academy of Quantum Molecular Science accepts the results of theoretical calculations, which are very near to those of real or experimental calculations.
Spatial arrangement of valeraldehyde
The molecule contains 5 carbon atoms, 1 simple oxygen atom which shows the presence of a formyl group, 10 hydrogen atoms and 1 double bond - 16 atoms in total (Figure 1). The molecular formula is C5H10O and SMILES (simplified molecular input line entry system) notation is CCCCC=O. The structure has no stereoisomers. Its molecular weight is 86 g/mol and estimated dipole moment is 2.4D.
Computational method
The molecular structure of valeraldehyde was drawn with the help of Avogadro 1.2.12 Then geometry of the molecule was optimised by applying the universal force field. Verification of theoretical results was done through KnowItAll Informatics Systems, an online system that compares results with previous deposits. We selected GAMESS (General Atomic and Molecular Electronic Structure System) for computational calculation. The simulation program MestreNova was used for prediction of carbon and hydrogen nuclear magnetic resonance (NMR) and Gabedit 2.50 was used to visualise the results. All the software programs used are free for academic use.
Parameter use
For determination of electronic spectra, calculation frequency in basic set STO-3G with Becke three-parameter Lee Yang-Parr (B3LYP) hybrid function code was used. This method increases the time for computation but produces better results. Parameters for theoretical Raman and UV-VIS calculations were the same.
Computational results
Most of the quantum chemical results for molecular vibrations of molecules with more than a few atoms were described by the Born-Oppenheimer approximation. According to the Born-Oppenheimer assumption, the electronic and nuclear motion of molecules can be treated separately.13 Computational scientists apply different computational capabilities to modelling and simulation, data analysis, and visualisation of results in their exploration of molecules. The successful outcome of the theoretical calculation is used as a reference for future work.
IR theoretical value
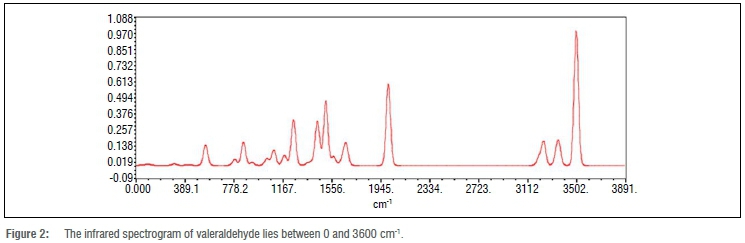
Figure 2 shows the IR spectrogram of valeraldehyde. The IR spectrogram is widely used for measurement, quality control, dynamic measurement and identification of the functional group. In the group frequency region of the spectrogram, peaks can be seen which correspond to functional groups.
From the IR spectrogram of valeraldehyde, for identification of the aldehyde group, formyl group stretching and C-H stretching, two sharp peaks of moderate intensity are observed near 2941 cm-1 and 2720 cm-1. But from the theoretical calculation, the peak is observed at 2007 cm-1.
C=O stretching vibration: 1725-715 cm-1 (strong)
C-H deformation vibration: 900-700 cm-1 (limited diagnostic value).
UV-Vis theoretical value
As can be seen in Figure 3, the UV-Vis spectrogram of valeraldehyde shows a parabolic curve. There are many applications of UV-Vis spectrophotometric studies. In this study, we used the UV-Vis spectrogram to examine electronic energy transition and excitation, that is, any molecule raised to a higher electron level means that an electron has been shifted from an orbital of lower energy to an orbital of higher energy
Raman theoretical value
Figure 4 shows the Raman spectrogram of valeraldehyde. Presence of the aldehyde group is confirmed by medium C-H stretching in the functional group region at 3735 cm-1.

Raman spectroscopy also reveals the molecular structure. Rotational and vibrational motions are unique attributes of every molecule, analogous to the fingerprints of humans. Each atom of the molecule is connected by a bond and each bond requires a different frequency, which creates different types of peaks - this region of the IR spectrum is called the fingerprint region.
As mentioned previously, aldehydes play a vital role in beverage production and determine the aqueous solution. Raman spectrophotometry is a superior tool for aqueous solutions because water can absorb the IR rays and thus distort the results of IR spectroscopic investigations. The Raman spectrogram is therefore widely used to complement the IR results.
NMR prediction
NMR provides information on the structure, dynamics and reaction state. NMR also confirms the three-dimensional structure at a molecular level and thus limits the need for X-ray crystallography. The overriding goal of NMR is to produce magnets with higher field strengths in an effort to gain sensitivity and chemical shift dispersion for measurement. Three NMR analyses were needed for confirmation of the structure of the valeraldehyde molecule: one for the presence of carbon (Figure 5a), one for the presence of hydrogen (Figure 5b) and one for the presence of oxygen (Figure 5c), as the molecular formula is C5H10O.
NMR is a tool used in the food industry for quality control and research. As mentioned above, aldehydes are a main food ingredient and foodomics requires assessment of food. IR and Raman spectroscopy provide information on structure only, whereas NMR provides information on the chemical composition of food, allowing sources of variation to be identified.
Electronic properties
Table 1 shows the band gap of valeraldehyde. The band gap or energy gap indicates the difference between energy at the ground state and energy at the first excited state and is calculated by subtracting the energy in electron volts (eV) at the lowest unoccupied molecular orbital (LUMO) from that at the highest occupied molecular orbital (HOMO).
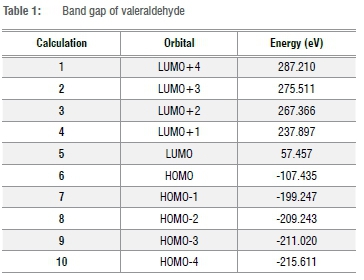
For valeraldehyde, the minimum calculated energy gap between LUMO and HOMO is 164.892 eV, which means that it is a poor electrical conductor and acts more like an insulator.
Aldehyde C-H bond dissociation energy is much smaller. The direct influence of electronegativity due to the hydrogen CO bond stabilises any negative intermediate. Here the essential gap is the distinction of electron affinity and upright ionisation potential. A narrow energy gap indicates a tranquil electron transition and a wide energy gap confers high thermodynamic solidness of the compound. As indicated by Koopmans' hypothesis, with regard to Hartree-Fock estimations, ionisation potential and electron affinity can be considered (short) energies of HOMO and LUMO, individually. At a presumption, the relation between band gap, which is classically associated with Fermi level to conduction band gap, and the HOMO-LUMO gap may be significant. An adverse feature of obtaining results from a molecular assessment (HOMO-LUMO) and a continuous model assessment of the solid state band gap is that it neglects unlike results. Unfortunately, these two models meet up somewhat badly in the nanodomain, because neither interpretation is very good in this crossover domain, giving rise to a possible new frontier area for research.
Thermochemistry
Thermochemical data provide information on the stability and reactivity of molecules. The enthalpy of the valeraldehyde molecule is -704086.66 kJ/mol, at a temperature of 25 °C and pressure of 1 atm. This result indicates that the molecule is exothermic in nature.
Conclusion
The current computational analysis is useful to predict even a complex aldehyde precursor and this approach may be useful for the assessment of other aldehyde molecule polymers. The resultant IR and Raman spectra of the compound were compared with the standard compound. Extra peaks in the spectrogram are indicative of impurities present in the compound. The field of computational analysis is still not fully developed enough to determine the vibrational property of organic molecules. The success of this theoretical approach opens a pathway to applying a correct algorithm force field for the assignment to aldehyde family. The key benefit of computational vibration analysis is an edge in the ease with which the electron shift, donation and withdrawing effects can be observed. The choice of open-source software allows for easier reproducibility of the methods. More recently, aldehyde nanomaterials are under development.14 Further, it is necessary to calculate the polarisation density, the magnitude of the displacement vector, the dipole moment, the dielectric constant, the electric susceptibility, and the refractive index for a complete description of electrical and optical properties of alkanes.
Acknowledgements
We thank Muhammad Yunus and Sohail Nadeem for a layout of the manuscript.
Authors' contributions
M.Az.: Sample analysis, writing the initial draft. M.An.: Supervision. S.I.: data validation.
References
1.Kubisa P, Neeld K, Starr J, Vogl O. Polymerization of higher aldehydes. Polymer. 1980;21(12):1433-1447. https://doi.org/10.1016/0032-3861(80)90145-7 [ Links ]
2.El Mubarak MA, Danika C, Vlachos NS, Farsalinos K, Poulas K, Sivolapenko G. Development and validation of analytical methodology for the quantification of aldehydes in e-cigarette aerosols using UHPLC-UV. Food Chem Toxicol. 2018;116:147-151. https://doi.org/10.1016/j.fct.2018.04.021 [ Links ]
3.Gibson B, Aumala V, Heiniö R-L, Mikkelson A, Honkapää K. Differential evolution of Strecker and non-Strecker aldehydes during aging of pale and dark beers. J Cereal Sci. 2018;83:130-138. https://doi.org/10.1016/j.jcs.2018.08.009 [ Links ]
4.Amending and correcting Annex II to Regulation (EC) nº 1333/2008 of the European Parliament and of the Council as regards the use of certain food additives. Off J Eur Union. 2013;L129(438):28-33. [ Links ]
5.Kuykendall JR, Kuykendall NS. Aldehydes. In: McQueen CA, editor. Comprehensive toxicology. 3rd ed. Oxford: Elsevier; 2018. p. 352-388. [ Links ]
6.Pelucchi M, Namysl S, Ranzi E, Frassoldati A, Herbinet O, Battin-Leclerc F, et al. An experimental and kinetic modelling study of n-C4C6 aldehydes oxidation in a jet-stirred reactor. Proc Combust Inst. 2019;37(1):389-397. https://doi.org/10.1016/j.proci.2018.07.087 [ Links ]
7.Gutteridge PA, Turner J. Computer aided materials selection and design. Mater Design. 1982;3(4):504-510. https://doi.org/10.1016/0261-3069(82)90161-3 [ Links ]
8.Smith P, Rennie A. Computer aided material selection for additive manufacturing materials. Virtual Phys Prototyp. 2010;5(4):209-213. [ Links ]
9.Siesler HW. Vibrational spectroscopy. In: Reference module in materials science and materials engineering. Amsterdam: Elsevier; 2016. [ Links ]
10.Barone V. Computational spectroscopy. In: Reference module in chemistry, molecular sciences and chemical engineering. Amsterdam: Elsevier; 2013. [ Links ]
11.Liu Y, Feng J, Liu X, Wang J. Theoretical study with DFT on the mechanism of visible light-driven β-functionalization of aldehydes. Comput Theor Chem. 2018;1123:154-160. https://doi.org/10.1016/j.comptc.2017.11.021 [ Links ]
12.Hanwell MD, Curtis DE, Lonie DC, Vandermeersch T, Zurek E, Hutchison GR. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J Cheminform. 2012;4(1), 17 pages. https://doi.org/10.1186/1758-2946-4-17 [ Links ]
13.Oppenheimer MBJR. Zur Quantentheorie der Molekeln [On the quantum theory of molecules]. Annalen der Physik. 1927;389(20):457-484. German. [ Links ]
14.Chandiramouli R. Structural and electronic properties of germanane nanosheet upon molecular adsorption of alcohol and aldehyde molecules: DFT comparative analysis. J Mol Liq. 2017;242:571-579. [ Links ]
 Correspondence:
Correspondence:
Muhammad Aziz
Aziz1sh@Hotmail.com
Received: 13 Oct. 2018
Revised: 08 Apr. 2019
Accepted: 08 Apr. 2019
Published: 30 Sep 2019
FUNDING: None
EDITOR: Priscilla Baker


{kind=link}
{kind=link}