










Servicios Personalizados
Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkSouth African Journal of Science
versión On-line ISSN 1996-7489
versión impresa ISSN 0038-2353
S. Afr. j. sci. vol.115 no.3-4 Pretoria mar./abr. 2019
http://dx.doi.org/10.17159/sajs.2019/4876
RESEARCH LETTER
A bioinformatics pipeline for rare genetic diseases in South African patients
Maryke SchoonenI; Albertus S. SeyffertII; Francois H. van der WesthuizenI; Izelle SmutsIII
IHuman Metabolomics, North-West University, Potchefstroom, South Africa
IICentre for Space Research, North-West University, Potchefstroom, South Africa
IIIDepartment of Paediatrics and Child Health, Steve Biko Academic Hospital, University of Pretoria, Pretoria, South Africa
ABSTRACT
The research fields of bioinformatics and computational biology are growing rapidly in South Africa. Bioinformatics pipelines play an integral part in handling sequencing data, which are used to investigate the aetiology of common and rare diseases. Bioinformatics platforms for common disease aetiology are well supported and continuously being developed in South Africa. However, the same is not the case for rare diseases aetiology research. Investigations into the latter rely on international cloud-based tools for data analyses and ultimately confirmation of a genetic disease. However, these tools are not necessarily optimised for ethnically diverse population groups. We present an in-house developed bioinformatics pipeline to enable researchers to annotate and filter variants in either exome or amplicon next-generation sequencing data. This pipeline was developed using next-generation sequencing data of a predominantly African cohort of patients diagnosed with rare disease.
Significance:
•We demonstrate the feasibility of in-country development of ethnicity-sensitive, automated bioinformatics pipelines using free software in a South African context.
•We provide a roadmap for development of similarly ethnicity-sensitive bioinformatics pipelines
Keywords: computational tools; African cohort; next-generation sequencing; rare disease
Introduction
The research fields of bioinformatics and computational biology are both growing rapidly in South Africa, with an ever-increasing number of both small and large laboratories having access to next-generation sequencing (NGS) technologies. This increase in sequencing capability continues to stimulate the development of a variety of platforms, databases and initiatives, such as H3Africa (https://www.h3africa.org), the South African Human Genome Programme (http://sahgp.sanbi.ac.za), and the South African Bioinformatics Institute (https://www.sanbi.ac.za), to support sequencing data analysis. However, to date, the majority of these developments have been focused on common disease related research, because diseases like HIV, fibromyalgia, tuberculosis and malaria are major health challenges in southern Africa.1
Southern African researchers doing rare-disease related research (diseases affecting 6-8% of the global population)2 still rely on flexible, international cloud-based tools, such as Bystro3, BrowseVCF4, and RD-Connect5, as their analysis needs have not yet been fully met locally. For European populations, these tools allow researchers to leverage well-established genotype-phenotype correlations to guide investigations into rare disease aetiology. However, these correlations do not always hold for African populations, thus significantly reducing the power of and degree to which these online tools can be relied on in the South African research context. A niche therefore exists for a tool that is both sensitive to population heterogeneity and general enough in nature to enable effective domestic rare disease research.
African population groups are heterogeneous with large genetic variety and limited information on genotype-phenotype correlations.6 Even though data are processed using the same reference genome, some aspects, such as allele frequency for disease-causing variants and genotype-phenotype correlation, could differ between ethnically diverse population groups7 and must be taken into account.
In this paper, we present a bioinformatics pipeline developed in-house to address these limitations. This pipeline processes NGS data of ethnically diverse population groups without any strong prior assumptions regarding genotype-phenotype correlations. This offline pipeline, suitable for the analysis of both exome and amplicon sequencing data, is written in Bash (or the Bourne-Again SHell) and uses only open-source software. To allow other researchers to benefit from this work, the pipeline has been made available on GitHub under the GNU GPLv3 software licence.8 The Ensembl Variant Effect Predictor (VEP)9 offline script is used for variant annotation, and the Genome Mining (GEMINI)10 command line database management tool for variant filtering. The pipeline is easily adjustable with regard to what annotations are made, and how they are filtered, which is especially useful when working with NGS data from ethnically diverse patients. The bioinformatics pipeline for variant annotation and filtering of amplicon and exome sequencing data presented here has been successfully used in research forming part of the project 'Investigating the aetiology of South African paediatric patients diagnosed with mitochondrial disorders'.11-14
Bioinformatics pipeline
Workflows leveraging NGS technology mainly consist of two parts: primary and secondary analysis. Figure 1 shows what this workflow looks like when our newly developed pipeline is incorporated. During primary analysis, patient samples are prepared and sequenced on a specialised platform such as Ion Torrent (used in our research case) or Illumina. These platforms further perform the requisite signalcalling, basecalling, reference sequence alignment and variant calling. The output from this primary analysis, for a single sample, is a Variant Call Format (VCF) file listing all the variants for that sample (variants_patientX.vcf.gz in Figure 1).15 Secondary analysis, often with the identification of disease-causing variants in mind, entails variant annotation and filtering. Variant annotation is typically done using purpose-built tools, such as ANNOVAR16 and VEP runner (used in our research case) that annotate variants with relevant metadata such as variant type, variant allele frequency and predicted impact. Variant filtering, in which variants of interest are identified, can be done using tools like BrowseVCF, RD-Connect and GEMINI (used in our research case). These tools filter variants based on the metadata associated with each, which allows a researcher to find the variants most relevant to their investigation.
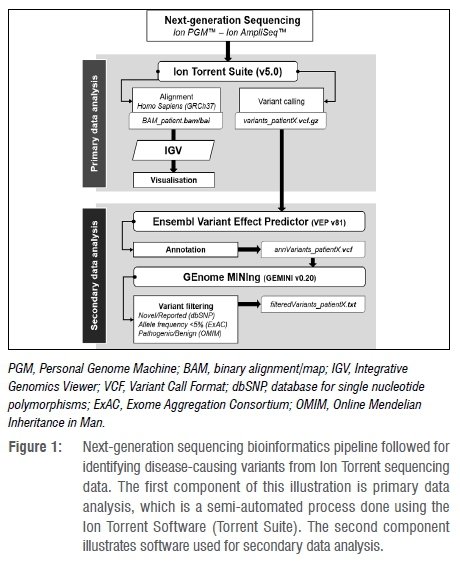
The pipeline presented in this paper is focused exclusively on secondary analysis because the problematic potential population biases, which stem from variant annotations made based on European-population-centric research results, influence only the secondary analysis results. The pipeline utilises the offline VEP script's flexibility to annotate variants sufficiently comprehensively so that population-sensitive variant filtering can be achieved using GEMINI's high-specificity querying capabilities. Our pipeline is intended to form part of a larger NGS data analysis workflow, and is only responsible for variant annotation and filtering.
Secondary data analysis
Bash is the default command shell on most modern Unix-like systems (and Unix-like tools for Windows), and incorporates useful features from the Korn shell and the C shell.17,18 The ability for Bash users to write powerful scripts to automate analysis has made it a popular choice for research implementations, typically in the form of command line tools and analysis pipelines. Many of these command line tools are available as free software, giving researchers who use the Bash shell access to a plethora of bioinformatics tools with which to do their research.
The main advantage of using Bash is that it allows powerful automation of established tools in a natural way while minimising both the number of introduced software dependencies and the need for more advanced analysis infrastructure. These are important advantages considering the rare-disease focus of this pipeline. For researchers who focus exclusively on rare diseases, whose expertise and research interests often lie outside bioinformatics, a pipeline based on Bash allows flexible analyses that remain easily repeatable over long periods of time, while necessitating minimal skills development and infrastructure investment.
Here, we present the bioinformatics pipeline developed using these tools during our research. Scripts were run on Slackware v14.12 with GNU Bash, version 4.4.12.
As primary analysis, using the Ion Torrent system, delivers sequencing results in batches, it seemed prudent to write the secondary analysis scripts to operate in a batch fashion. This approach, coupled with the built-in automation that comes with scripting, ensures consistency across the samples in each batch and across different batches.
In the secondary analysis pipeline, the scripts implement two steps: variant annotation and data mining. Variant annotation is done using the VEP script, which can be downloaded from Ensembl's website.9 Data mining is done using the command line tool GEMINI, which uses the SQLite relational database management system to enable effective sequencing data mining.10 Each of these steps has a Bash script dedicated to it: vep_single.sh and gemini_single.sh, respectively (the '.sh' extension indicates that these text files are Bash scripts). These scripts are embedded into the vep_batch.sh and gemini_batch.sh scripts, respectively, which run them for each .vcf.gz file in a given directory. The hetero_annotate.sh script binds these two batch scripts into a full pipeline, calling each in sequence and managing their inputs and outputs. See Appendices 1-6 in the supplementary material for the full contents of these scripts. A brief description of what each script does is given below.
The vep_single.sh script (Supplementary appendix 1) takes an Ion Torrent-generated input .vcf.gz file as its first argument, and runs the VEP for it. It takes an output file as its second argument (a .vcf file), to which the annotated output of the VEP is written. Alongside these two arguments, a number of additional arguments are passed to the VEP script, with the most notable being the --fields [list] argument, which allows for the specification of the required annotation fields included in the annotated output .vcf file. This argument (as well as the specification of the VEP arguments) is handled in an extensible way by the vep_single.sh script, and the list of desired fields can be built up over multiple lines, or in multiple groups. A list of all possible fields and a list of all possible additional arguments can be found in the VEP's documentation.9,19 The VEP also generates a 'statistic run report' (.html file) when run, containing general statistics that give information on, among other things, the number of variants processed, number of overlapping genes, and number of novel/reported variants.
The gemini_single.sh (Supplementary appendix 2) script takes a VEP-annotated input .vcf file as its first argument and loads it into a SQLite database (a .db file). This file is then mined for relevant variants using user-defined SQLite database query specifications. Each line in the queries_spec.txt (Supplementary appendix 3) auxiliary text file contains one such query specification, and consists of two comma-separated fields: the query's name and the relevant SQLite query snippet. Queries can easily be added to or removed from this file, or a different such file can be specified in the gemini_single.sh script. An example of such a query, which filters based on variants' allele frequency in African populations, is: rareAFR,aaf_1kg_afr <= 0.01.
What information these queries should return for each variant is controlled by the cols variable, which is used to build the full SQLite query. Some examples of columns that can be included in a query are is_coding (which is true if the variant is in a coding region), rs_ids (which lists the rsIDs associated with each variant), and aaf_1kg_afr (which stores the allele frequency of the variant in the AFR population as reported in the 1000 Genomes Project). For a list of all possible columns, see GEMINI's documentation.20 The cols variable is handled similarly to the --fields [list] argument to the VEP, and is similarly extensible. The output of each query is a list of variants, with information from the database columns specified in the cols variable, stored in a .txt file that has the query's name as filename. These files constitute the main output of the pipeline.
Once all the queries have been performed, a meta_info.txt file is generated that summarises the lengths of the lists contained in the query output files. This file, along with the generated query outputs, is saved in the folder specified as the second argument to the gemini_single.sh script.
The vep_batch.sh and gemini_batch.sh scripts (Supplementary appendices 4 and 5, respectively) each run their counterpart script, as discussed above, for all the .vcf.gz files in a given directory. Both of these scripts take the directory containing the relevant input files as their first argument, with the second argument being the directory to which output should be written.
Finally, the hetero_annotate.sh script (Supplementary appendix 6) administers the operation of the two batch scripts described above. The first argument of this script is the directory containing the Ion Torrent output .vcf.gz files (the first argument for the vep_batch.sh and vep_single.sh scripts), and the second argument is the directory to which the GEMINI output should be written (the second argument to the gemini_batch.sh and gemini_single.sh scripts).
From here the user can further prioritise and filter the variants according to the criteria of their choice. For our African data set, variants were first filtered based on the novelty of these variants. Second, variants were filtered based on African allele frequencies, as reported in Exome aggregation consortium (ExAC21). In addition, for mostly non-African population groups, known disease-causing variants can be identified from the data set using databases such as the Online Mendelian Inheritance in Man (OMIM22) and ClinVar23.
Conclusion
Robust bioinformatics pipelines are key components for diagnosis and research of rare genetic diseases. Here we describe an offline, flexible and open-source bioinformatics pipeline that annotates variants using VEP and filtering of important disease-causing variants using GEMINI from NGS data. It was developed as a first prudent step towards data processing and offers unique advantages for the detection of multiple genetic alterations, including in patients of African descent for whom little information is available. The pipeline can be used on exome as well as amplicon NGS data and was designed using NGS data of a predominantly African cohort with rare disease. Identifying underlying genetic causes for rare disease is particularly challenging in the South African population, with limited bioinformatics support for researchers and non-bioinformaticians. With the increased burden to diagnose rare genetic diseases using NGS and genetic screening, and the limited support and resources in developing countries such as South Africa, it is equally important to develop and provide access to bioinformatics pipelines, such as this one. With continued development, pipelines in South Africa could be further refined and made more user-friendly, making them useful for both researchers and clinicians. These refinements could include, for instance, cloud-based interfaces like those used in developed countries. With such refinements in place, clinicians would more easily be able to investigate genotype-phenotype correlation in rare diseases for African population groups.
Acknowledgements
We acknowledge the Medical Research Council of South Africa for financial support in funding the overarching research project titled 'Investigating the aetiology of South African paediatric patients diagnosed with mitochondrial disorders'.
Authors' contributions
M.S. and A.S.S. were responsible for: conceptualisation, methodology and writing - the initial draft. F.H.v.d.W. was responsible for: writing - revisions, student supervision and project leadership. I.S. was responsible for: writing - revisions, student supervision and funding acquisition.
References
1.Mulder NJ, Christoffels A, De Oliveira T, Gamieldien J, Hazelhurst S, Joubert F, et al. The development of computational biology in South Africa: Successes achieved and lessons learnt. PLoS Comput Biol. 2016;12(2), e1004395, 15 pages. https://doi.org/10.1371/journal.pcbi.1004395 [ Links ]
2.Dawkins HJ, Draghia-Akli R, Lasko P, Lau LP, Jonker AH, Cutillo CM, et al. Progress in rare diseases research 2010-2016: An IRDiRC perspective. Clin Transl Sci. 2018;11(1):11-20. https://doi.org/10.1111/cts.12501 [ Links ]
3.Kotlar AV, Trevino CE, Zwick ME, Cutler DJ, Wingo TS. Bystro: rapid online variant annotation and natural-language filtering at whole-genome scale. Genome Biol. 2018;19(1), Art. #14, 11 pages. https://doi.org/10.1186/s13059-018-1387-3 [ Links ]
4.Salatino S, Ramraj V. BrowseVCF: A web-based application and workflow to quickly prioritize disease-causative variants in VCF files. Brief Bioinform. 2016;18(5):774-779. https://doi.org/10.1093/bib/bbw054 [ Links ]
5.Thompson R, Johnston L, Taruscio D, Monaco L, Béroud C, Gut IG, et al. RD-Connect: An integrated platform connecting databases, registries, biobanks and clinical bioinformatics for rare disease research. J Gen Intern Med. 2014;29(3):780-787. https://doi.org/10.1007/s11606-014-2908-8 [ Links ]
6.Van der Westhuizen FH, Sinxadi PZ, Dandara C, Smuts I, Riordan G, Meldau S, et al. Understanding the implications of mitochondrial DNA variation in the health of black southern African populations: The 2014 Workshop. Hum Mutat. 2015;36(5):569-571. https://doi.org/10.1002/humu.22789 [ Links ]
7.Shearer AE, Eppsteiner RW, Booth KT, Ephraim SS, Gurrola II J, Simpson A, et al. Utilizing ethnic-specific differences in minor allele frequency to recategorize reported pathogenic deafness variants. Am J Hum Genet. 2014;95(4):445-453. https://doi.org/10.1016/j.ajhg.2014.09.001 [ Links ]
8.Seyffert AS. A set of BASH scripts for annotating and filtering next-generation sequencing data using Ensembl's offline VEP runner GEMINI [software]. c2018 [cited 2018 Oct 01]. Available from: https://github.com/aseyffert/hetero_annotate [ Links ]
9.McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GR, Thormann A, et al. The Ensembl variant effect predictor. Genome Biol. 2016;17(1), Art. #122, 14 pages. https://doi.org/10.1101/042374 [ Links ]
10.Paila U, Chapman BA, Kirchner R, Quinlan AR. GEMINI: Integrative exploration of genetic variation and genome annotations. PLoS Comput Biol. 2013;9(7), e1003153, 8 pages. https://doi.org/10.1371/journal.pcbi.1003153 [ Links ]
11.Van der Westhuizen FH, Smuts I, Honey E, Louw R, Schoonen M, Jonck L-M, et al. A novel mutation in ETFDH manifesting as severe neonatal-onset multiple acyl-CoA dehydrogenase deficiency. J Neurol Sci. 2017;384:121-125. https://doi.org/10.1016/j.jns.2017.11.012 [ Links ]
12.Louw R, Smuts I, Wilsenach K-L, Jonck L-M, Schoonen M, Van der Westhuizen FH. The dilemma of diagnosing coenzyme Q10 deficiency in muscle. Mol Genet Metab. 2018;125(1-2):38-48. https://doi.org/10.1016/j.ymgme.2018.02.015 [ Links ]
13.Van der Walt EM, Smuts I, Taylor RW, Elson JL, Turnbull DM, Louw R, et al. Characterization of mtDNA variation in a cohort of South African paediatric patients with mitochondrial disease. Eur J Hum Genet. 2012;20(6):650-656. https://doi.org/10.1038/ejhg.2011.262 [ Links ]
14.Smuts I, Louw R, Du Toit H, Klopper B, Mienie LJ, Van der Westhuizen FH. An overview of a cohort of South African patients with mitochondrial disorders. J Inherit Metab Dis. 2010;33(3):95-104. http://dx.doi.org/10.1007/s10545-009-9031-8 [ Links ]
15.Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, et al. The variant call format and VCFtools. Bioinformatics. 2011;27(15):2156-2158. https://doi.org/10.1093/bioinformatics/btr330 [ Links ]
16.Wang K, Li M, Hakonarson H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38(16), e164, 7 pages. https://doi.org/10.1093/nar/gkq603 [ Links ]
17.Ramey C, editor. Bash, the Bourne−Again Shell. In: Proceedings of The Romanian Open Systems Conference & Exhibition (ROSE 1994), The Romanian UNIX User's Group (GURU); 1994 November 3-5; Bucharest, Romania. [ Links ]
18.Stevens WR, Rago SA. Advanced programming in the UNIX environment. Ann Arbor, MI: Addison-Wesley; 2013. [ Links ]
19.The Ensembl Genome Database Project, NCBI, NIH. Variant Effect Predictor [software]. c2018 [cited 2018 Oct 01]. Available from: https://www.ensembl.org/info/docs/tools/vep/index.html [ Links ]
20.Genome Mining. GEMINI: A flexible framework for exploring genome variation [software]. c2017 [cited 2018 Oct 01]. Available from: https://gemini.readthedocs.io/en/latest/ [ Links ]
21.Karczewski KJ, Weisburd B, Thomas B, Solomonson M, Ruderfer DM, Kavanagh D, et al. The ExAC browser: Displaying reference data information from over 60 000 exomes. Nucleic Acids Res. 2016;45(D1):D840-D845. https://doi.org/10.1093/nar/gkw971 [ Links ]
22.Amberger JS, Bocchini CA, Schiettecatte F, Scott AF, Hamosh A. OMIM.org: Online Mendelian Inheritance in Man (OMIM®), an online catalog of human genes and genetic disorders. Nucleic Acids Res. 2014;43(D1):D789-D798. https://doi.org/10.1093/nar/gku1205 [ Links ]
23.Landrum MJ, Lee JM, Riley GR, Jang W, Rubinstein WS, Church DM, et al. ClinVar: Public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2013;42(D1):D980-D985. https://doi.org/10.1093/nar/gkt1113 [ Links ]
 Correspondence:
Correspondence:
Maryke Schoonen
mschoonen28@gmail.com
Received: 18 Apr. 2018
Revised: 01 Nov. 2018
Accepted: 03 Dec. 2018
Published: 27 Mar. 2019

