










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSouth African Journal of Science
On-line version ISSN 1996-7489
Print version ISSN 0038-2353
S. Afr. j. sci. vol.114 n.3-4 Pretoria Mar./Apr. 2018
http://dx.doi.org/10.17159/sajs.2018/20170324
RESEARCH ARTICLE
Screening of the NIH Clinical Collection for inhibitors of HIV-1 integrase activity
Shaakira AbrahamsI, II; Salerwe MosebiI; Muhammed Q. FishI, II; Maria A. PapathanasopoulosII; Raymond HewerIII
ICentre for Metal-Based Drug Discovery, Advanced Materials Division, Johannesburg, South Africa
IIDepartment of Molecular Medicine and Haematology, University of the Witwatersrand Medical School, Johannesburg, South Africa
IIIDiscipline of Biochemistry, University of KwaZulu-Natal, Pietermaritzburg, South Africa
ABSTRACT
Drug repurposing offers a validated approach to reduce drug attrition within the drug discovery and development pipeline through the application of known drugs and drug candidates to treat new indications. Full exploitation of this strategy necessitates the screening of a vast number of molecules against an extensive number of diseases of high burden or unmet need and the subsequent dissemination of the findings. In order to contribute to endeavours within this field, we screened the 727 compounds comprising the US National Institutes of Health (NIH) Clinical Collection through an HIV-1 (human immunodeficiency virus type 1) integrase stand transfer inhibition assay on an automated scintillation proximity assay platform. Only two compounds were identified within the initial screen, with cefixime trihydrate and epigallocatechin gallate found to reduce integrase strand transfer activity at IC50 values of 6.03±1.29 μM and 9.57±1.62 μM, respectively. However, both cefixime trihydrate and epigallocatechin gallate retained their low micromolar inhibitory activity when tested against a raltegravir-resistant integrase double mutant (FCIC50 values of 0.83 and 0.06, respectively), were ineffective in an orthogonal strand transfer ELISA (<30% inhibition at 100 juM) and produced negligible selectivity index values (<1) in vitro. While no useful inhibitors of HIV-1 integrase strand transfer activity were found within the NIH Clinical Collection, the identification of two assay-disrupting molecules demonstrates the importance of consideration of non-specific inhibitors in drug repurposing screens.
SIGNIFICANCE:
• This study is the first to screen the US NIH Clinical Collection for potential HIV-1 integrase inhibitors.
• The pervasive nature of promiscuous inhibitors is emphasised.
Keywords: drug repurposing; strand transfer; pan-assay interference compounds; PAINS
Introduction
Early-stage drug discovery fulfils a critical role within the broader drug discovery process and the entire drug discovery and development pipeline. Early-stage drug discovery is typically - but not always - undertaken following target identification and validation, and involves the screening of compounds with the intent purpose of identifying compounds with promising activity (HIT compounds) that can then be developed further (into LEAD compounds) within the drug discovery phase. Early-stage drug discovery activities can range from the evaluation of a limited set of compounds, typically selected through rational drug design methodologies, to the assessment of large compound libraries through high throughput screening (HTS; defined as the screening of >10 000 compounds per day) and even ultra-HTS (uHTS; defined as the screening of >100 000 compounds per day) operations. Owing to the sheer number of compounds screened, early-stage drug discovery ostensibly carries the highest failure rate and, accordingly, the highest risk of all activities within the pipeline. However, the true bottleneck to success in the broader drug discovery and development process lies less with the quantity of compounds identified as HITS during screening and more with the quality of these compounds and their suitability as drug candidates. Specifically, the highest cause for compound attrition in the pipeline, by far, is attributed to non-clinical toxicity which accounts for the termination of >40% of all compounds from the drug discovery and development pipeline.1
To mitigate the potential significant financial loss resulting from compound failures, in particular the high cost of late-stage failures, most pharmaceutical organisations adopt the 'fail early, fail cheap' paradigm. To support this approach, researchers aim to recognise ADMET-related issues through an ever-increasing number of tests undertaken at progressively earlier stages of the pipeline. Equally, findings from these tests have been retrospectively accumulated to delineate physiochemical properties (i.e. Logp LogD, molecular weight, aromatic rings, rotatable bonds, polar surface area, etc.) that influence drug-likeness and then subsequently collated into 'rules of thumb' (such as the Lipinski rule of five, the rule of three and many other variations and extensions) and property prediction software programs. Application of these predictive models has allowed for the identification and judicious removal of non-favourable compounds either following screening or directly from the physical compound library prior to screening. While immeasurably useful, these tools have not proven infallible as evinced through a recent study of 812 failed compounds (oral development candidates from four different major pharmaceutical companies) that could draw no correlation between non-clinical toxicology failure and physiochemical properties.1 Similarly, a subset of compounds eliciting growing interest because of their subversive effects in drug discovery efforts are promiscuous inhibitors2,3 or pan-assay interference compounds (PAINS)4. These compounds yield convincing false-positive results in biological assays and significant efforts have been undertaken to identify them and ultimately remove them from screening libraries.4 Broadly categorised and inclusive of several classes of compounds with varying mechanisms of action (i.e. aggregate-inducing compounds, redox-cyclers, covalent modifiers, metal complexes), these compounds do not readily lend themselves to predictive algorithms. Nonetheless, databases of existing PAINS highlight common structures (i.e. flavonoids, quinones, rhodamines) and some can also be searched for similarity.
Of other approaches aimed at minimising compound attrition, the concept of drug repurposing (or drug repositioning) has drawn significant interest. Herein, the underlying principle is the evaluation of clinically approved drugs or late-stage clinical trial failures (all off-patent or generics) as disease-modifying agents in therapeutic areas other than the one for which they were designed or proved effective. The main appeal of this approach is the decreased risk of failure arising from safety issues while the extensive prior development allows for a quicker transitioning through the pipeline (up to 60%) with reduced costs (up to 40%). If successful, the drug can be granted patent protection on grounds of a new application or new formulation. The classic example of drug repositioning is Viagra® - the blockbuster erectile dysfunction drug from Pfizer which first served as an angina medication under the name Sildenafil. Numerous other examples exist, including azidothydimine (the cancer turned anti-HIV drug), ropinirole (a dual Parkinson's and restless legs syndrome treatment) and Rogaine® (a hair-loss drug repurposed from a blood pressure drug from a failed ulcer candidate), to mention but a few. The scientific merit of the concept has driven growth in its popularity as is clearly evident through interest from major pharmaceutical companies, the growth in focused start-up companies, the rise in related literature and a recently launched journal (Drug Repurposing, Rescue and Repositioning) with dedicated content.
Applying the drug repurposing approach to the field of HIV drug discovery has been previously described. While the treatment options for HIV-1 are formidable - both in the number of antiretroviral agents approved and the efficacy of combination therapy - the absence of an effective therapeutic vaccine or cure and the persistent threat of antiretroviral drug resistance has substantiated the continued exploration for novel inhibitors. In this study, we sought to identify an existing drug with activity against HIV-1 integrase (IN) - a virally encoded enzyme that catalyses the integration of viral DNA into the host chromosome. For this purpose we screened the US National Institutes of Health (NIH) Clinical Collection (NCC), which is a 727 small-molecule library of FDA-approved and late-stage candidates that has been previously explored for proteasome stimulators5 and coronavirus inhibitors6 but not, to the best of our knowledge, for HIV-1 IN inhibition. The NCC library was screened by means of an automated process through an HIV-1 IN strand-transfer (ST) inhibition scintillation proximity assay (SPA) in order to identify novel catalytic IN inhibitors.
Methods
Expression and purification of recombinant HIV-1 integrase
The reagent pINSD.His (Cat. #2957) was obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH, from Dr Robert Craigie.78 Recombinant His-tagged HIV-1 IN was expressed and purified as previously described.9 Briefly, wild-type HIV-1 IN was overexpressed in E. coli BL21 (DE3) bacterial cells using the NL4-3 histidine (HIS)-tagged HIV-1 IN coding sequence, pINSD, cloned into pET15B (Merck Millipore, Darmstadt, Germany). Cells were grown to logarithmic phase in Luria-Bertani medium and induced with 1 mM isopropyl-thio-galactoside. The recombinantly expressed protein was purified through affinity chromatography using a nickel (Ni)-affinity column and buffer exchanged into storage buffer (20 mM HEPES pH 7.2, 1 M NaCl, 4 mM EDTA, 2 mM dithiothreitol and 50% glycerol) using a PD-10 Sephadex column (GE Healthcare, Buckinghamshire, UK). The expression and purification of the HIV-1 IN was confirmed through SDS-PAGE and subsequent Western blot analysis. Similarly, recombinant IN which incorporated raltegravir-resistant mutations, INQ148H/G140S, was prepared. Briefly, the pINSD.His plasmid was used as a template for mutagenesis with the QuickChange Lightning Site Directed Mutagenesis kit (Agilent Technologies, Santa Clara, CA, USA). Raltegravir-resistant mutations were inserted into the pINSD.His backbone and included Q148H/ G140S substitutions.
Radiolabelling of target DNA for scintillation proximity assays
Annealed oligonucleotides for target DNA (tDNA), T 56-S (AAAAGGAGGA-GAAGGAAAGGAGAGAGAGCGAATTAGCCCTTGGTC) and T 56-A (AAAAGGA GGAGAAGGAAAGGAGAGAGAGGACCAAGGGCTAATTCG) oligonucleotides (Inqaba Biotech, Pretoria, South Africa), were radiolabelled with 3H-dCTP and 3H-dTTP (AEC Amersham, Johannesburg, South Africa) by filling in the 5'-overhangs according to the Fermentas Klenow fragment DNA labelling kit instructions (ThermoFisher Scientific, Waltham, MA, USA). Unincorporated nucleotides were removed from the radiolabelled tDNA using the QIAquick nucleotide removal kit (Qiagen, Hilden, Germany).
HIV-1 integrase strand transfer scintillation proximity assay
The SPA was carried out as previously described10 and adapted to an automated platform on a Hamilton Starlet robotic system (Hamilton, Bonaduz, Switzerland). Briefly, a 10x reaction buffer was prepared containing 200 mM HEPES (pH 7.5), 300 mM NaCl, 50 mM dithiothreitol and 0.5% Igepal (nonidet-P40). Polyvinyltoluene streptavidin-coated scintillation beads (GE Healthcare Sciences, Marlborough, MA, USA) were reconstituted in 1x reaction buffer at a final concentration of 10 mg/ mL. Biotinylated donor DNA (dDNA) was added at a final concentration of 500 nM and rocked at room temperature for 1 h. The bead suspension was washed twice with 1x reaction buffer and centrifuged at 1000 x g for 5 min. The pellet was resuspended at 2 mg/mL in 2x reaction buffer to which recombinant IN (wild-type or Q148H/G140S mutant) was added at a final concentration of 1 uM and rocked at room temperature for 30 min. The final SPA reactions comprised, per well: 1 mg/mL SPA bead-dDNA-IN complex with 8 to 10 test compounds at 10 μM each for single-dose experiments or concentrations ranging from 100 to 0.78 uM for dose-response experiments (substituted with DMSO buffer solution for blank control). This reaction mixture was incubated at 22 °C for 30 min whilst shaking gently. The reactions were initiated by adding 500 nM 3H-tDNA to each well at a final concentration of 50 nM and incubated at 37 °C shaking for 90 min before the enzymatic reaction was stopped using 62 mM EDTA. The reaction product formation was measured using the Top Count Scintillation Counter NXT (Perkin Elmer, Waltham, MA, USA). Percentage inhibition was determined for single-dose experiments while IC50 values were determined as the compound concentration required to reduce HIV-1 recombinant IN ST activity by 50% and calculated using OriginPro 8.0 software (Origin Lab Corporation, Northampton, MA, USA). All inhibition values are the average of at least triplicate experiments.
HIV-1 integrase strand transfer enzyme-linked immunosorbent assay
The HIV-1 IN strand transfer inhibition enzyme-linked immunosorbent assay (ELISA) was adapted from previously described methods.9,11 Briefly, 0.15 μM double-stranded biotinylated dDNA (5'-biotin-ACCCTTTTAGTCAGTGT GGAAAATCTCTAGCA-3' and 5'-ACTGCTAGAGATTTTCCACACTGACTAA AAG-3') was added to the wells of streptavidin-coated 96-well microtitre plates (R&D Systems, Minneapolis, MN, USA). Following incubation at room temperature for 60 min and a stringent wash step, 1 uM purified recombinant HIV-1 subtype B IN (in the presence of MgCl2) was assembled onto the pre-processed dDNA through incubation for 30 min at 22 °C. Following a wash step, the test compounds were titrated into individual wells at a final concentration of 100 uM for single-dose evaluation or concentrations ranging from 100 to 0.78 uM for dose-response experiments. The microtitre plates were incubated for 30 min at 37 °C, washed and the strand transfer reaction was initiated through the addition of 0.25 uM double-stranded FITC-labelled target DNA (5'-TGACCAAGGGCTAATTCACT-FITC-3' and 5'-AGTGAATTAGCCCTTGGTCA-FITC-3') in Hepes buffer containing MgCl2 and MnCl2. After an incubation period of 60 min at 37 °C, the plates were washed as before and an alkaline phosphatase conjugated anti-FITC secondary antibody (Sigma-Aldrich, St Louis, MO, USA) was added. Finally, the plates were washed and substrate (BluePhos, KPL, Gaithersburg, MD, USA) was added to allow for detection at 620 nm using a microplate reader (xMarkTM, Bio-Rad, Hercules, CA, USA). Percentage inhibition was determined for single-dose experiments while IC50 values were determined as the compound concentration required to reduce HIV-1 recombinant IN ST activity by 50% and calculated using OriginPro 8.0 software (Origin Lab Corporation). All inhibition values are the average of at least triplicate experiments.
Cytotoxicity assays
The reagent MT-4 (Cat. #120) was obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH, from Dr Douglas Richman.12-14 The cytotoxicity assay was performed as per standard methods and as described previously.9,15 Briefly, MT-4 cells were plated in 96-well microtitre plates at 3.0 x 105 cells/mL and allowed to stabilise for 2 h at 37 °C and 5% CO2. Thereafter, test compounds were added to the plate through twofold serial dilution to allow for eight final compound concentrations ranging from 200 to 1.56 uM in a total volume of 200 uL/ well. The cells and compounds were then incubated for 96 h at 37 °C and 5% CO2. To each well, 20 uL CellTiter 96 Aqueous One Solution (Promega, Madison, WI, USA) was added. The plates were incubated for 4 h and absorbance was read at 490 nm on a multiplate reader (xMark, Bio-Rad). CC50 values were determined as the concentration of the test compound required to reduce the cell viability by 50% and were calculated using OriginPro 8.0 software (Origin Lab Corporation). The values obtained are averages of at least three separate experiments.
Antiviral activity
To determine antiviral activity, 50 uL HIV-1NL4-3 virus was added to 3.0x105 MT-4 cells/mL at a multiplicity of infection of 0.1 and the mixture was spinoculated at 3000 x g for 90 min. After washing off unbound virus, cells were plated in 96-well microtitre plates at 100 uL/ well and allowed to stabilise for 1 h at 37 °C and 5% CO2. Thereafter, test compounds were added to the plate through twofold serial dilution to allow for eight final compound concentrations ranging from 200 to 1.56 uM in a total volume of 200 uL/well. The cells and compounds were then incubated for 96 h at 37 °C and 5% CO2. Cell-free supernatants were collected from each well, and p24 concentration was determined using the Vironostika HIV-1/2 p24 Antigen ELISA (bioMerieux, Marcy-I 'Etoile, France) as per manufacturer's instructions. EC50 values were determined as the concentration of the test compound required to reduce p24 concentration by 50% and were calculated using OriginPro 8.0 software (Origin Lab Corporation). The values obtained are averages of at least three separate experiments. Selectivity index (SI) values were calculated as the ratio of CC50/EC50.
Results and discussion
Inhibition of recombinant integrase strand transfer activity
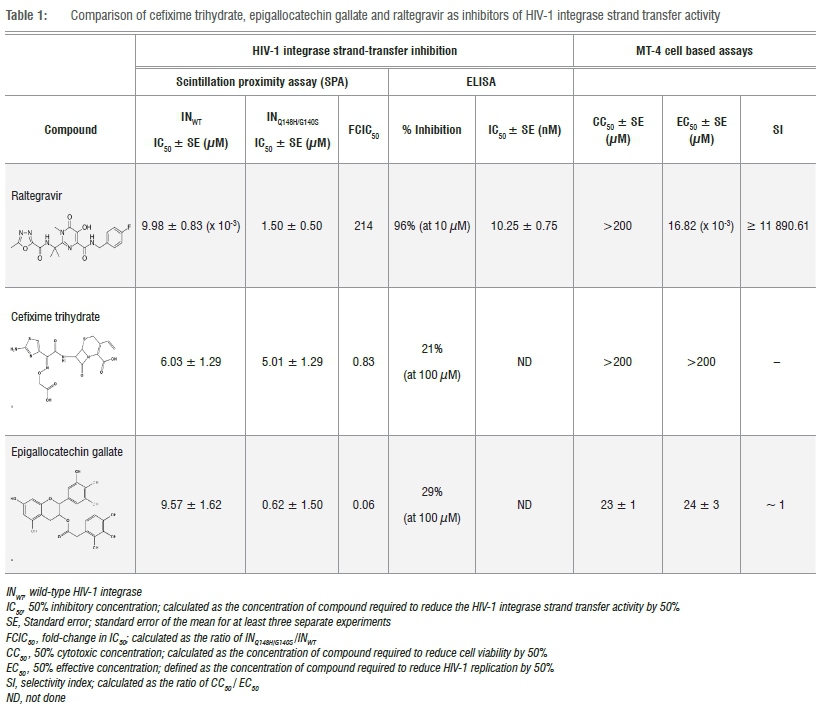
To begin the screening process, all 727 small molecules within the NCC library were pooled in an orthogonal manner into 171 pools; each pool comprised a combination of 8 or 10 different compounds with each compound present in two pools. The pools were then screened through an automated SPA, in triplicate, with each compound evaluated at a final single-dose concentration of 10 uM. Initially, 24 pools were found to be affected by colour-quenching which was only resolved through the identification and removal of 12 coloured compounds. On completion of the screening process, four pools were found to reduce recombinant INWT ST activity by the pre-defined minimum cut-off of >50%, indicating the presence of two active compounds. Through cross-referencing, the two common compounds were identified as cefixime trihydrate (CEF) and epigallocatechin gallate (EGCG). CEF, a third-generation orally administered cephalosporin is a potent, broad-spectrum bactericidal. Like other cephalosporins, CEF possesses a dihydrothiazine ring fused to a beta-lactam ring and derives its therapeutic effect through inhibition of cell-wall synthesis by disruption of the transpeptidation process. Modulation of HIV-1 replication by CEF has not been previously described in the literature; however, cephalosporin oligonucleotides and monocyclic ^-lactams have been reported as HIV-1 protease inhibitors. EGCG is a polyphenolic acid ester that has been proclaimed as an effective agent for an exceedingly broad range of diseases (including HIV-1 through several distinct mechanisms of action) despite its well-documented promiscuous nature.4,16 A search of the US NIH registry and results database of clinical studies revealed 92 clinical trials in varying stages of completion (from recruiting to completed) examining EGCG as treatment for 276 different clinical conditions.17
In our experience, the HIV-1 integrase ST SPA is a robust assay that yields a low number of HIT compounds per screen. The inclusion of detergent within the protocol design serves, perhaps inadvertently, to reduce the identification of false positive responses caused by promiscuous molecules. We therefore opted to continue investigating EGCG to verify a non-specific mechanism, and as such, both CEF and EGCG were subjected to dose-response evaluation within the previously described SPA. Here we established IC50 values of 6.03±1.29 uM and 9.57±1.62 uM for CEF and EGCG, respectively (Table 1) with steep slopes observed for both compounds (Hill slope > 1). In comparison, raltegravir, the first-in-class HIV-1 integrase drug marketed as Isentress®, inhibited recombinant INWT activity by 92±5% at a single-dose concentration of 10 uM and produced an IC50 of 9.98±0.83 nM (Table 1) with a Hill slope of 1. Thereafter, the compounds were tested for inhibitory activity of the raltegravir-resistant INQ148H/G140S double-mutant.18 Replication capacity of this double mutant was reduced to 59% of INWT and, as anticipated,19 raltegravir was significantly less effective against this mutant (p<0.01) as reflected by a fold change in IC50 (FCIC50) value of 214 (Table 1). On the contrary, the two identified compounds retained their micromolar inhibitory activity against the raltegravir-resistant double mutant with FCIC50 values of 0.83 and 0.06 calculated for CEF and EGCG, respectively (Table 1). As a further direct evaluation of activity, the compounds were tested for ST inhibition within an HIV-1 IN ST assay based on an ELISA platform. As a true, indisputable IN ST inhibitor, raltegravir efficiently inhibited INWT in this orthogonal assay to the same degree as that observed within the SPA-based assay (IC50 = 10.25±0.75 nM) while dose-response evaluations were not warranted for CEF and EGCG as both proved ineffective inhibitors at a high single-dose concentration (<30% inhibition at 100 uM; Table 1). Up to this point, the behaviour of CEF and EGCG strongly supported a nonspecific role for both molecules through a related mechanism of action that disrupted the SPA-based platform. This observation subsequently led us to the work of another group that speculated a non-specific mechanism for CEF within an SPA - in this case as an inhibitor of both NADH dehydrogenase (at an IC50 ~ 8 μM) as well as MurG (at an IC50 ~ 16 μM).20
In-vitro evaluation
EGCG and CEF were both evaluated for toxicity within the MT-4 mammalian cell line. While EGCG yielded a CC50 of 23 uM, CEF was not found to be toxic within the limits of the assay (>200 uM). Thereafter, inhibition of HIV-1 replication by CEF and EGCG was evaluated in vitro in the MT-4 cell line following infection by HIV-1 NL4-3. For EGCG, an EC50 of 24 uM was determined through dose-response studies (Table 1). The observed viral inhibition by EGCG closely correlated the toxicity profile of the compound in the same cell line and led to a negligible SI value Table 1). An SI value for CEF could not be determined as no observable viral inhibition was found in the cell-based HIV-1 inhibition assay up to the maximum tested compound concentration of 200 uM (Table 1). As a control, raltegravir was found to inhibit HIV-1 replication within this assay with an EC50 of 16.82 nM with no observable toxicity up to the limit of the assay (CC50 > 200 uM; SI value > 11 890.61).
Conclusion
Drug repurposing has proven successful in the past and offers a viable strategy for the discovery and development of therapeutic agents. In an endeavour to contribute to efforts in this field, we screened the NCC library to identify new inhibitors of HIV-1 integrase strand transfer activity. While no true inhibitors of HIV-1 IN ST activity were discovered, the identification of two non-specific inhibitors through our screen demonstrated that drug repurposing is not insusceptible to the presence of assay disruptors. In particular, the identification of EGCG demonstrates the invasiveness of even the most well-documented PAINS into chemical screening libraries. Furthermore, and perhaps more interestingly, the confirmation of the clinically relevant antibiotic CEF as an SPA disruptor demonstrates necessity to interrogate the action of well-characterised molecules within specific assay platforms and also supports the mandatory use of secondary or orthogonal assays to confirm inhibition. The findings from this study suggest that both EGCG and CEF disrupt the SPA through a similar non-aggregating mechanism that will be elucidated through future studies to facilitate further screening projects based on this assay platform.
Acknowledgements
We thank the National Research Foundation of South Africa, Mintek, the University of the Witwatersrand and the University of KwaZulu-Natal for funding and permission to publish these findings.
Authors' contributions
R.H. was responsible for the conceptualisation, methodology, data analysis, validation, critically reviewing the writing, writing revisions, student supervision, project leadership, project management and funding acquisition. S.M. was responsible for the methodology, data analysis, validation, data curation, writing revisions, student supervision and project leadership. S.A. was responsible for the methodology, data collection, data analysis, sample analysis, validation, data curation and writing the initial draft. M.Q.F. was responsible for the methodology, data collection and data analysis. M.A.P was responsible for the methodology, data analysis, validation, writing revisions and student supervision.
References
1. Waring MJ, Arrowsmith J, Leach AR, Leeson PD, Mandrell S, Owen RM, et al. An analysis of the attrition of drug candidates from four major pharmaceutical companies. Nat Rev Drug Discov. 2015;14:475-486. https://doi.org/10.1038/nrd4609 [ Links ]
2. McGovern SL, Caselli E, Grigorieff N, Schoichet BK. A common mechanism underlying promiscuous inhibitors from virtual and high-throughput screening. J Med Chem. 2002;45:1712-1722. https://doi.org/10.1021/jm010533y [ Links ]
3. McGovern SL, Helfand BT, Feng B, Shoichet BK. A specific mechanism of nonspecific inhibition. J Med Chem. 2003;46:4265-4272. https://doi.org/10.1021/jm030266r [ Links ]
4. Baell J, Walters MA. Chemistry: Chemical con artists foil drug discovery. Nature. 2014;513(7519):481-483. https://doi.org/10.1038/513481a [ Links ]
5. Trader DJ, Simanski S, Dickson P Kodadek T. Establishment of a suite of assays that support the discovery of proteasome stimulators. Biochim Biophys Acta. 2017;1861(4):892-899. https://doi.org/10.1016/j.bbagen.2017.01.003 [ Links ]
6. Cao JJ, Forrest C, Zhang X. A screen of the NIH Clinical Collection small molecule library identifies potential anti-coronavirus drugs. Antiviral Res. 2015;114:1-10. https://doi.org/10.1016/j.antiviral.2014.11.010 [ Links ]
7. Bushman FD, Engelman A, Palmer I, Wingfield P, Craigie R. Domains of the integrase protein of human immunodeficiency virus type 1 responsible for polynucleotidyl transfer and zinc binding. Proc Natl Acad Sci USA. 1993;90:3428-3432. https://doi.org/10.1073/pnas.90.8.3428 [ Links ]
8. Craigie R, Hickman AB, Engelman A. Integrase. In: Karn J, editor. HIV. Volume 2: A practical approach. Oxford: Oxford University Press; 1995. p. 53-71. [ Links ]
9. Harrison AT, Kriel FH, Papathanasopoulos MA, Mosebi S, Abrahams S, Hewer R. The evaluation of statins as potential inhibitors of the LEDGF/p75 - HIV-1 integrase interaction. Chem Biol Drug Des. 2014;85(3):290-295. https://doi.org/10.1111/cbdd.12384, [ Links ]
10. Grobler JA, Stillmock KA, Hazuda DJ. Scintillation proximity assays for mechanistic and pharmacological analyses of HIV-1 integration. Methods. 2009;49(4):249-253. https://doi.org/10.1016/j.ymeth.2009.03.002 [ Links ]
11. Hazuda DJ, Hastings JC, Wolfe AL, Emini EA. A novel assay for the DNA strand-transfer reaction of HIV-1 integrase. Nucleic Acids Res. 1994;22:1121-1122. https://doi.org/10.1093/nar/22.6.1121 [ Links ]
12. Harada S, Koyanagi Y Yamamoto N. Infection of HTLV-III/LAV in HTLV-I-carrying cells MT-2 and MT-4 and application in a plaque assay. Science. 1985;229:563-566. https://doi.org/10.1126/science.2992081 [ Links ]
13. Larder BA, Darby G, Richman DD. HIV with reduced sensitivity to zidovudine (AZT) isolated during prolonged therapy. Science. 1989;243:1731-1734. https://doi.org/10.1126/science.2467383 [ Links ]
14. Pauwels R, De Clercq E, Desmyter J, Balzarini J, Goubau P, Herdewijn P, et al. Sensitive and rapid assay on MT-4 cells for detection of antiviral compounds against the AIDS virus. J Virol Meth. 1987;16:171-185. https://doi.org/10.1016/0166-0934(87)90002-4 [ Links ]
15. Mphahlele MK, Papathanasopoulos MA, Cinellu MA, Coyanis EM, Mosebi S, Traut T, et al. Modification of HIV-1 reverse transcriptase and integrase activity by gold(III) complexes in direct biochemical assays. Bioorg Med Chem. 2012;20:401-407. https://doi.org/10.1016/j.bmc.2011.10.072 [ Links ]
16. Ingolfsson HI, Thukur P Herold KF, Hobart EA, Ramsey NB, Periole X, et al. Phytochemicals perturb membranes and promiscuously alter protein function. ACS Chem Biol. 2014;9(8):1788-1798. https://doi.org/10.1021/cb500086e [ Links ]
17. ClinicalTrials.gov. A service of the U.S. National Institutes of Health [homepage on the Internet]. No date [cited 2017 Mar 20]. Available from: https://clinicaltrials.gov [ Links ]
18. Malet I, Delelis O, Valantin MA, Montes B, Soulie C, Wirden M, et al. Mutations associated with failure of raltegravir treatment affect integrase sensitivity to the inhibitor in vitro. Antimicrob Agents Chemother. 2008;52(4):1351-1358. https://doi.org/10.1128/AAC.01228-07 [ Links ]
19. Witmer M, Danovich R. Selection and analysis of HIV-1 integrase strand transfer inhibitor resistant mutant viruses. Methods. 2009;47:277-282. https://doi.org/10.1016/j.ymeth.2009.02.025 [ Links ]
20. Ravishankar S, Kumar VP, Chandrakala B, Jha RK, Solapure SM, De Sousa SM. Scintillation proximity assay for inhibitors of Escherichia coli MurG and, optionally, MraY Antimicrob Agents Chemother. 2005;49:1410-1418. https://doi.org/10.1128/AAC.49.4.1410-1418.2005 [ Links ]
 Correspondence:
Correspondence:
Raymond Hewer
Email: hewerr@ukzn.ac.za
Received: 21 Sep. 2017
Accepted: 12 Nov. 2017
Published: 27 Mar. 2018
FUNDING: National Research Foundation (South Africa)


{kind=link}