










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSouth African Journal of Science
On-line version ISSN 1996-7489
Print version ISSN 0038-2353
S. Afr. j. sci. vol.111 n.11-12 Pretoria Nov./Dec. 2015
http://dx.doi.org/10.17159/sajs.2015/20140417
RESEARCH ARTICLE
In-vitro effects of protease inhibitors on BAX, BCL- 2 and apoptosis in two human breast cell lines
Gbenga A. AdefolajuI, II; Katherine E. TheronIII; Margot J. HosieI, III
ISchool of Anatomical Sciences, University of the Witwatersrand, Johannesburg, South Africa
IIDepartment of Medical Sciences, Public Health and Health Promotion, University of Limpopo, Polokwane, South Africa
IIINewcastle University Medicine, Johor, Malaysia
ABSTRACT
Currently, the treatment of choice of HIV/AIDS in South Africa is the multidrug combination regimen known as HAART (highly active antiretroviral treatment). HAART, which commonly consists of nucleoside or non-nucleoside reverse transcriptase inhibitors and protease inhibitors, has radically decreased mortality and morbidity rates among people living with HIV/AIDS. The emphasis of the original development of the antiretroviral drugs was on clinical effectiveness (reducing mortality). Presently, emphasis has shifted from the initial short- term considerations to the long-term undesirable or harmful effects induced by this treatment regimen. Whether antiretroviral compounds are oncogenic is widely speculated, which led to this investigation into the effects of protease inhibitors on the expression of key apoptotic regulatory genes, BAX and BCL-2, in two human breast cell lines, MCF-7 and MCF-10A by real-time qPCR gene expression and immunofluorescence. The anti-apoptotic effects of the protease inhibitors - LPV/r were also investigated by cell death detection ELISA and acridine orange staining. This study also evaluated the cytotoxicity of the antiretroviral drugs in normal and cancer cell lines of the breast (at clinically relevant concentrations of the drugs and at different time points, 24-96 h), employing the neutral red uptake assay. The drugs and combinations tested did not alter BAX and BCL-2 gene expression and protein expression and localisation in both cell lines. In addition, the protease inhibitors-LPV/r did not inhibit camptothecin-induced apoptosis in both cell lines. We have shown that the protease inhibitors demonstrated varying degrees of cytotoxicity in the breast cells. The resulting DNA damage associated with cytotoxicity is strongly implicated in the processes of tumour initiation.
Keywords: lopinavir; ritonavir; apoptosis; breast cancer
Introduction
The HIV/AIDS pandemic has had a catastrophic effect on the world population. In 2011, approximately 34 million people were recorded as living with HIV globally. Sub-Saharan Africa, which is home to just 10% of world's population contains a huge 69% of people living with HIV or AIDS.1 South Africa has the highest number of people living with HIV in sub-Saharan Africa and in the world.1 It is for this reason that in November 2003, South Africa started the world's largest public sector rollout of highly active antiretroviral treatment (HAART) to date.2
HAART has radically decreased mortality and morbidity among people living with HIV and AIDS3,4,5 The emphasis of the original development of the antiretroviral (ARV) drugs was on clinical effectiveness (reducing mortality) with all other considerations secondary.6 Presently, emphasis has shifted from the initial short-term considerations to the long-term undesirable or harmful effects induced by this treatment regimen.6 Whether antiretroviral compounds are oncogenic remains to be fully elucidated.7 Torres et al.8 suggest that the clinical use of nucleoside/nucleotide reverse transcriptase inhibitor (NRTI) drug pairs may lead to additive or synergistic effects compounding long-term risk for cancer gene mutations and potential carcinogenesis.
Breast cancer is the most common cancer seen in women worldwide.9 HIV-infected patients on antiretroviral treatment are reported to survive longer due to the treatment and therefore are at danger of developing breast cancer because its risk increases with age.9,10,11 Other studies suggest that the use of ARV drugs might increase breast cancer risk by fat redistribution from peripheral and gluteal tissues to the breast as a result of increased production of oestrogen.9 The total influence of HAART on the risk for non-AIDS defining malignancies (NADM) is far from concluded. In a meta-analysis,12 the standardised incidence ratio of some non-AIDS defining malignancies, including breast cancer, was substantially increased prior to and in the HAART era. In a Swiss HIV cohort study, in which a mega prospective cohort of people were tracked for an average of about 5 years after HAART no definite influence of HAART on standardised incidence ratios of NADMs was revealed.13 However, another HIV/AIDS cohort conducted in England7, showed that a substantial total rise in risk for NADMs objectively accompanied HAART treatment. Non-AIDS defining cancers now generally account for more deaths in HIV-infected individuals than AIDS defining cancers. Although traditional risk factors may account for some of this discrepancy, they certainly do not explain the explosion of these cancers in HIV-affected people on HAART14 Engels et al.15 found that non-AIDS defining cancers comprise 58% of all cancer deaths post-HAART (1996-2002) in comparison to 31.4% in the pre- HAART era (1991-1995). These data strongly suggests that antiretroviral drugs may influence cancer development and progression.
In many cancers, there is a consistent pattern of apoptosis inhibition and deregulation of cell and tissue homeostasis. Numerous studies implicate apoptosis-related genes and their products in the development of cancer.16-18 The induction of apoptosis occurs via two major pathways: the extrinsic (death receptor) pathway and the intrinsic (mitochondria) pathway.16 The extrinsic pathway is activated by the attachment of the Fas plasma-membrane death receptor (and related receptors like tumour necrosis receptor 1 and its family) to its extracellular ligand, Fas-L. The intrinsic pathway also leads to apoptosis but under the control of mitochondrial pro-enzymes. The majority of cellular death in vertebrates follows the mitochondrial pathway of apoptosis.17 This pathway to cell death is controlled by the B-cell CLL/lymphoma 2 (BCL-2) family of proteins. They operate to regulate the integrity of the mitochondrial outer membrane. When apoptosis arises as a result of cooperation among these proteins, the two pro-death BCL-2 effector proteins; BCL-2-associated X protein (BAX) and BCL-2 antagonistic killer (BAK), disrupt this membrane in a process known as mitochondrial outer membrane permeabilisation.17 If mitochondrial outer membrane permeabilisation takes place, proteins located in the mitochondrial inter-membrane compartment enter the cytosol and activate caspases and cysteine proteases that coordinate the disassembling of the cell.17 The process of mitochondrial outer membrane permeabilisation is opposed by the pro-survival BCL-2 proteins, such as BCL-2, BCL-W and BCL-xL, which impedes the capacity of BAX and Bak to permeabilise. The relevance of this impact in cancer is highlighted by the remark that oncogenes, like Myc, which advances proliferation, also encourage cell death that is inhibited by the anti-apoptotic BCL-2 proteins.17 The widely studied genes associated with apoptosis include BCL-2; the anti-apoptotic gene and BAX; a pro-apoptotic gene.18 Hypothetically BCL-2 over-expression renders a survival advantage for cancer cells; in contrast, BAX stimulation restores sensitivity to apoptosis induced by drugs or radiation.18 In this study, we hypothesised that HIV protease inhibitors might inhibit the pro-apoptotic BCL-2 family member BAX, or activate the anti-apoptotic BCL-2 family member thereby promoting cell survival and lead to cancer development and/or progression.
In the majority of published studies, the in-vitro effects of antiretroviral drugs on immune cells were investigated. Few studies on non-immune cells were found. In the current study, we therefore investigated the effects of some of the drugs in the South African HAART treatment guidelines, individually and in combination, on BAX and BCL-2 gene expression in two breast cell lines at clinically relevant concentrations. BAX and BCL-2 proteins were localised with immunofluorescence. We also determined whether the protease inhibitors - LPV/r, reported to have anti-apoptotic effects,19-20 can inhibit drug-induced apoptosis in these non-immune cell lines. We also evaluated the cytotoxicity of the antiretroviral drugs in normal and cancer cell lines of the breast at clinically relevant concentrations of the drugs and at different time points (24 h, 48 h, 72 h and 96 h) by employing the neutral red uptake assay.
Materials and methods
Cells
Human breast carcinoma MCF-7 cells (American Type Culture Collection, Rockville, MD, USA) were cultured in DMEM medium (Gibco BRL, Gaithersburg, MD, USA) containing 10% foetal bovine serum (Gibco).
Immortalised human breast epithelial cells, MCF-10A (American Type Culture Collection) were cultured in mammary epithelium growth medium (MEGM; Lonza, Walkersville, MD, USA) containing human recombinant epidermal growth factor, hydrocortisone, insulin and bovine pituitary extract (Lonza).
Cells were cultivated as a stationary monolayer in plastic tissue-culture dishes (Nunclon, Denmark) and were incubated at 37 °C in a 5% CO2 humidified environment. The cells were incubated with the drugs individually and in combination at the indicated physiologically relevant concentrations for 24-96 h. The normal breast MCF-10A cells were cultured in serum-free media. MCF-7 cells were synchronised by serum starvation for 24 h21,22 prior to all experiments. After being cultured in complete medium for overnight attachment, MCF-7 cells were incubated with serum-free media for 24 h. The cells were then treated with the antiretroviral drugs (as described in Table 1) in medium supplemented with serum for the indicated periods.
This study was approved by the Human Research Ethics Committee of the University of the Witwatersrand (Clearance Certificate reference: M10905).
Drugs and treatment
The drugs were administered at clinically relevant concentrations, which reflect their steady-state plasma peak concentration.23,24 Lopinavir (LPV) and ritonavir (RTV) were purchased from Toronto Research Chemicals (Toronto, Ontario, Canada) and were dissolved in methanol. The cells were incubated with the drugs individually and in combination at the indicated concentrations (Table 1) for 24 h, 48 h, 72 h and 96 h. Normal and vehicle control groups were exposed to growth medium and vehicle, respectively (Groups 1 and 2).
The cells were exposed to 9.8 μg/mL LPV (Group 3) and LPV/r (Kaletra® Group 5). Even though the absolute bioavailability of ritonavir had not been determined in humans at the time of this study25, across studies, daily administration of Kaletra® 400/100 mg yields mean steady-state lopinavir plasma concentrations 15-20 times higher than those of ritonavir in individuals living with HIV-1.26 Therefore a 9.8 μg/mL LPV:0.6 μg/mL RTV ratio was used. In Group 4, cells received 0.6 μg/mL RTV alone.
Kits, antibodies and reagents
The neutral red TOX-4 kit, methanol, acridine orange (AO), ethylenediamine-tetraacetic acid (EDTA) and camptothecin (CPT) were purchased from Sigma Chemical Co. (St. Louis, MO, USA). Rabbit polyclonal anti-BAX antibody and mouse monoclonal anti-BCL-2 were purchased from DAKO (DK-2600, Glostrup, Denmark). Secondary antibodies, goat anti-rabbit rhodamine-conjugated and goat anti-mouse FITC-conjugated were purchased from Abcam (Cambridge, UK). The GeneJET RNA Purification Kit, DNase I, RNase-free kit and O'GeneRuler Low Range DNA Ladder were purchased from Thermo Scientific (Pittsburgh, PA, USA). High Capacity cDNA Reverse Transcription Kit and Power SYBR® Green PCR Master Mix were purchased from Life Technologies (California, CA, USA). Oligos for quantitative polymerase chain reaction (qPCR) was purchased from Integrated DNA Technologies, Inc. (Coralville, IA, USA). Agarose D-1 Low EEO-GQT was purchased from Conda Laboratories (Madrid, Spain).
Neutral red uptake assay
The neutral red uptake cytotoxicity assay is a precise, easy and reproducible assay that allows for distinguishing between viable and non-viable cells.27 Neutral red is a weak cationic supravital dye that is actively transported across the undamaged cell membrane of viable cells and accumulates in the lysosomes. Alterations to the lysosomal membrane integrity arising from the toxicity of substances reduces neutral red uptake thereby allowing the distinction between viable and damaged cells.27 After the viable cells have incorporated the dye, the dye is subsequently liberated from the cells and the degree of cytotoxicity, quantified by a spectrophotometer, is a measure of how many cells excluded the dye.27 This assay was used to evaluate the cytotoxic effects of the protease inhibitors used in this study. MCF-7 and MCF-10A cells were cultured in the presence of the drugs, or diluent at the indicated concentrations for periods of time ranging from 24 h to 96 h. For each cell line, a 96-well Falcon tissue culture plate was seeded with cells in replicates for each group including negative and vehicle controls (Table 1). After 24 h, 48 h, 72 h and 96 h of treatment with antiretroviral drugs, the medium was removed and the manufacturer's protocol27 was followed for the assay. Cells were rinsed in phosphate buffered saline (PBS). Neutral red medium was added to each well and plates were incubated for 2 h at 37 °C for the neutral red dye to be taken up by viable cells. Thereafter, the neutral red solution was removed and cells were rinsed with PBS. The solubilising solution (1% acetic acid in 50% ethanol) was added to each well for 10 min in order to extract the dye. The absorbance of the extracted dye was measured in a microplate reader (Anthos 2010 Model 17-550, Salzburg, Austria) at a wavelength of 540 nm. Background absorbance was read at 690 nm and subtracted from the 540 nm measurement. All experiments were repeated three times on three different days.
Total RNA extraction, cDNA synthesis, and real-time qPCR analysis
Cells were exposed to the drugs for up to 96 h and total RNA extraction was performed using the GeneJET RNA Purification kit according to the manufacturer's instructions. RNA concentration and purity were determined by using the Nanodrop-1000 spectrophotometer. RNA integrity was checked by gel electrophoresis. According to the manufacturer's instructions, genomic DNA was removed from total RNA using the DNase I, RNase-free kit. The DNAase I treated RNA was again cleaned with the GeneJET RNA Purification kit, re-quantified and stored at -80 °C until used. According to the manufacturer's instructions, cDNA was synthesised using the MultiScribe™ Reverse Transcriptase from 700 ng RNA. The reverse transcriptase reaction was carried out in a GeneAmp® PCR System 9600 Thermal Cycler for 10 min at 25 °C, 120 min at 37 °C and then the enzyme was deactivated for 5 min at 85 °C.
The cDNA aliquots were then utilised in qPCR reactions for BAX and BCL-2, with TBP, RPLP0 and TFRC used as the endogenous reference genes. PCR reactions were amplified for 40 cycles prior to which the AmpliTaq Gold® DNA polymerase was activated for 10 min at 95 °C. Each cycle consisted of a denaturing step for 15s at 95 °C, and annealing/extension step for 1 min at 60 °C. PCR amplification was performed in a final volume of 20 using the Power SYBR® Green PCR Master Mix with the ABI 7500 real-time PCR machine. Primer sequences and PCR product sizes are indicated in Table 2. To confirm the absence of nonspecific amplification, PCR products were separated on 3% agarose gels, stained with ethidium bromide and images acquired with the BioRad Gel Doc® XR (Model 170-8170 Segrate, Milan, Italy). Melt curves were generated for each PCR product using the Applied Biosystems ABI 7500 software. The relative mRNA expression levels of target genes in each sample were calculated using the qbasePLUS software (Biogazelle, Zulte, Belgium). The expression stability of the reference genes was evaluated using qbasePLUS version 2.3. This software uses a pair-wise comparison model to calculate the stability of each reference gene, and selects the two or more most stable genes from a panel of reference genes for normalisation.28 Genes are ranked based on a gene stability parameter M, where a low M value indicates high expression stability. To further indicate how stable a gene is expressed, the qbasePLUS software also calculates a coefficient of variation. PCR baseline quantification cycle (Cq) values were exported from the ABI 7500 software as an Excel file (Microsoft, Redmond, WA, USA) and imported into the qbasePLUS software. The data was analysed with the default settings and the arithmetic mean of replicates was used. Data from standard curve experiments from the ABI 7500 software, imported into the qbasePLUS software, was used to generate amplification efficiencies and standard errors that are used downstream by the qbasePLUS software to determine normalised gene expression levels. The relative quantity of each target/sample combination was scaled to the average Cq of corresponding target (scale set to untreated control in the qbasePLUS software). The relative expression of specific genes in the experiment were normalised as a ratio to the amount of the two most stably expressed reference genes according to the -∆∆CT method of Livak and Schmittgen29.
Immunofluorescence of BAX and BCL-2
The ABCAM® double labelling procedure was followed for immuno-fluorescence analysis. 1x104 cells were plated on cover slips 1 day before the experiment. Cells were treated with the drugs at the indicated combinations and concentrations for up to 96 h. Following treatment, cells were washed three times with 0.5% BSA in PBS, followed by fixation in 10% phosphate buffered formalin for 10 min. The fixed cells were rinsed in PBS and permeabilised with 0.05% Triton-X 100, washed and blocked with 10% normal goat serum in PBS for 60 min to eliminate nonspecific binding of the secondary antibody. Cells were then incubated with polyclonal rabbit anti-human BAX (1:1000 DAKO) and monoclonal mouse anti-human BCL2 (1:100 DAKO) overnight at 4 °C in a moist chamber. Following overnight incubation, the cells were washed and incubated for 2 h with a FITC-conjugated goat anti-mouse antibody (1:500 ABCAM) and rhodamine-conjugated goat anti-rabbit antibody (1:1000 ABCAM) in the dark. Slides were rinsed, nuclei counterstained in 4',6-diamidino-2-phenylindole, dihydrochloride (DAPI) 300 nM for 5 min, rinsed and mounted with fluoromount (Sigma). Negative control groups were set up to ensure that the secondary antibodies were specific for their primary antibodies. The primary antibodies were substituted with 0.1% BSA/PBS and the normal protocol was carried out. In a second negative control, the cells were treated in the same way as the experimental slides, but the secondary antibodies were substituted with PBS. The positive controls used in this study were HeLa cells, which had been previously shown to express BAX and HepG2 cells, shown to express BCL-2.
Cells were visualised using a Zeiss Laser Scanning Confocal Microscope 780 under a Zeiss 100X oil immersion objective. Slides were kept dark once fluorescent antibodies had been added, to prevent bleaching. As fading of the fluorochromes will take place, images were analysed within the same time period. The image acquisition settings remained constant for all exposures. Images were taken and, using the ZEN 2010 (Carl Zeiss, Jena, Germany) image analysis software, the intensity of the fluorescence of each micrograph was analysed. This analysis was done by using the software to initially define the regions of interest - the nucleus and cytoplasm. The mean intensity of each region of interest from the treatment groups was then analysed with the statistics software JMP® (Version 10.0 SAS Institute Inc., Cary, NC, USA). Data are reported as mean+SEM. After verifying the normal distribution and the homogeneity of the variance using an F test (p<0.05), a one-way analysis of variance (where a significance level of p<0.05 was set) was used to compare the results.
Acridine orange staining
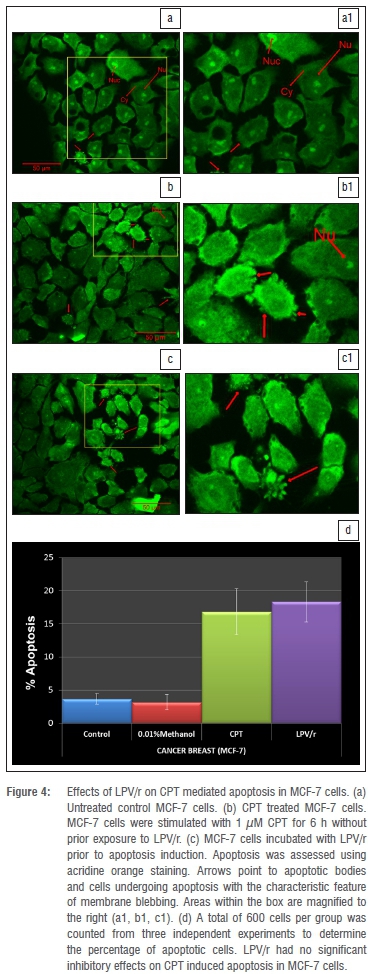
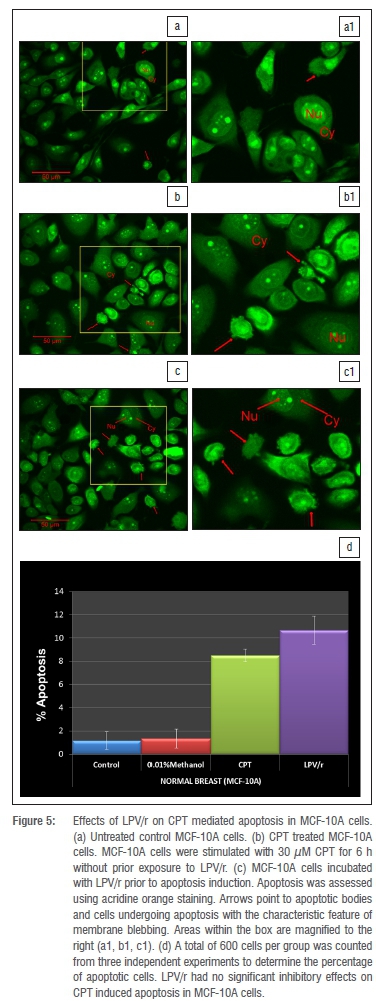
Fluorescence light microscopy with differential uptake of fluorescent DNA binding dyes such as AO provides a simple, rapid, and accurate method for measuring apoptosis and cell membrane integrity.30 Acridine orange permeates all cells, making the nuclei appear green. Live cells therefore have a normal green nucleus; early apoptotic cells have a bright green nucleus with condensed or fragmented chromatin; late apoptotic cells display condensed and fragmented chromatin and membrane blebbing.30,31 This procedure was carried out to determine whether the protease inhibitors inhibit drug-induced apoptosis in the breast cell lines. MCF-7 and MCF-10A cells were routinely grown on 22-mm square coverslips placed into 35-mm culture dishes (Costar, Cambridge MA, USA). Cells were treated with methanol and the protease inhibitor based combinations, LPV/r, for 96 h before being stimulated32 to undergo apoptosis with 1 CPT (MCF-7) and 30 CPT (MCF-10A) for 6 h. After induction of apoptosis, cells were stained with the AO dye mix for 5 min. The dye mix was 100 ^g/mL AO in PBS.33 After AO staining, culture dishes were inverted and fixed with formaldehyde vapour for 1 min to prevent the photo-damaging effects of continuous excitation on living cells because of the photosensitising effects of most fluorescent dyes.34 A total of 600 cells per cell line per group was counted from three independent experiments to determine the percentage of apoptotic cells.
Cell death detection
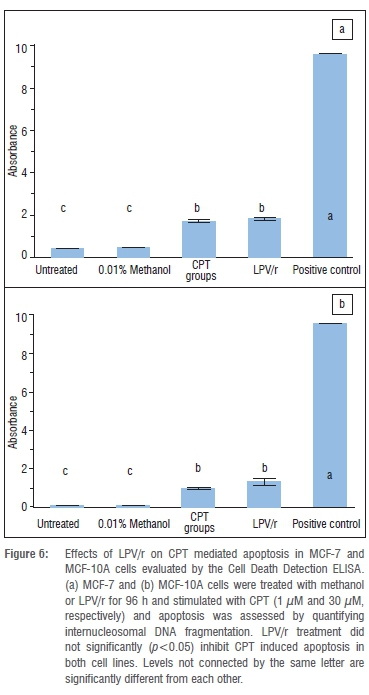
The Cell Death Detection ELISAplus kit (Roche Molecular Biochemicals, Mannheim, Germany) is used to measure changes in apoptosis.35,36 In this procedure, internucleosomal DNA fragmentation is quantitatively assayed by antibody mediated capture and detection of cytoplasmic mononucleosome and oligonucleosome associated histone-DNA complexes. Cells were cultured (as described) in 96-well plates in duplicate. Cells were treated with 0.01% methanol (vehicle) and the protease inhibitor based combination LPV/r (in the appropriate medium) for 96 h before being stimulated32 to undergo apoptosis with 1 μΜ CPT (MCF-7)37 and 30 μΜ CPT (MCF-10A)38 for 6 h.
According to the protocol described by Liu et al.35 and Tu et al.36, cells were centrifuged (200 g), resuspended in 200 μL of the lysis buffer supplied in the kit and incubated for 30 min at room temperature. Nuclei were then pelleted at 200 g for 10 min and 20 μL of the supernatant (cytoplasmic fraction) from the treated group, untreated group, positive control and background control were transferred into the corresponding streptavidin-coated wells for the enzyme-linked immunosorbent assay (ELISA) according to the manufacturer's standard protocol.36 Then, 80 μL of the immunoreagent was added to each well containing the 20 μL supernatant or controls. The wells were covered with adhesive foil and incubated on a microplate shaker under gentle shaking (300 rpm) for 2 h at room temperature. The solution was then removed by gentle pipetting; wells were rinsed three times with 250 μL incubation buffer and removed. Next, 100 μL of the ABTS solution was pipetted into the wells and incubated on a plate shaker at 250 rpm until the colour development was sufficient for photometric analysis (approximately 10-20 min). Then 100 μL of the ABTS stop solution was added to each well. Absorbance was measured at 405 nm and 490 nm (reference wavelength) with a microplate reader (Anthos 2010 Model 17-550). Signals in wells containing the substrate only were subtracted as background. Data were analysed from three independent experiments. Data analysis was performed using JMP® (Version 10.0). Data are reported as mean+SEM. After verifying the normal distribution and the homogeneity of the variance using an F test (p<0.05), a one-way analysis of variance (significance level p<0.05) was used to compare the results. If there was a significant difference between the means (p<0.05), then a Tukey-Kramer post-hoc analysis was performed to determine which treatment groups were significantly different from each other.
Statistical analysis
Statistical analysis was performed using JMP® (Version 10.0). Data are reported as mean+SEM. After verifying the normal distribution and the homogeneity of the variance using an F test (p<0.05), a one-way analysis of variance (where a significance level of p<0.05 was set) was used to compare the results. All means were then compared using the Tukey-Kramer honestly significant different test.
Results
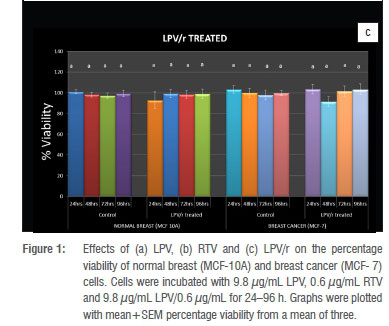
Effects of ARV drugs on the viability of MCF-7 and MCF 10A cells
The effects of the antiretroviral (ARV) drugs on the viability of MCF-7 and MCF-10A breast cells were investigated by the neutral red uptake assay. LPV significantly (p<0.05) reduced the viability of the normal breast MCF-10A cells at 24 h when compared to the vehicle control (Figure 1a). Also at 24 h, LPV significantly (p<0.05) reduced the percentage viability of the normal breast cells when compared to the cancer breast MCF- 7 cells (Figure 1a). With continuous exposure, the normal breast cells recovered from the initial cytotoxic effects of LPV, such that from 48 h to 96 h, there were no significant differences between the treated cells and their vehicle controls (Figure. 1a). LPV did not significantly (p<0.05) alter the percentage viability of the breast cancer MCF-7 cells from 24 h to 96 h when compared to their vehicle controls (Figure 1a The reduction in % viability of MCF-7 cells at 48 h was not statistically significant. RTV significantly (p<0.05) reduced the viability of the normal breast MCF-10A cells when compared to the vehicle control and the breast cancer MCF-7 cells at 24 h (Figure 1b). At 48 h, there was no significant (p<0.05) alteration in percentage viability between RTV treated and untreated in either cell line. At 72 h, RTV treated MCF-10A cells demonstrated significantly (p<0.05) increased viability, while at 96 h, there was no statistical significance in the difference between RTV treated and untreated MCF-10A cells (Figure 1b). RTV did not alter the percentage viability of the breast cancer MCF-7 cells from 24 h to 96 h (Figure 1b). The apparent increase in % viability of RTV treated MCF-7 cells at 24 h did not achieve statistical significance. The combination LPV/r interestingly did not significantly (p<0.05) alter the percentage viability of the normal (MCF-10A) and cancer (MCF-7) breast cells from 24 h to 96 h when compared to their respective vehicle controls and to each other at the respective time points (Figure 1c).


Effects of ARV drugs on BAX/BCL2 gene expression in MCF-7/MCF10A cells
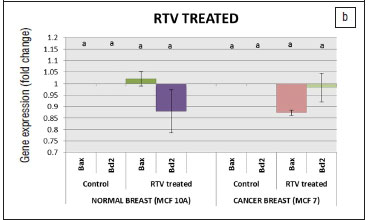
The effects of the protease inhibitors on BAX and BCL-2 mRNA expression in both cell lines were investigated after exposure for 24 h to 96 h (data shown for 96 h). In both cell lines treated with LPV, the differences in fold changes were not significantly (p<0.05) different from the untreated controls for BAX and BCL-2 (Figure 2a). In both cell lines treated with RTV and LPV/r, the differences in fold changes were not significantly (p<0.05) different from the untreated controls for BAX and BCL-2 (Figures 2b and 2c). Results were analysed with the qBASEPLUS software and normalised to the expression levels of TBP, TFRC and RPLP0.


Effects of ARVs on BAX/BCL2 immunofluorescence in MCF-7/10A cells
Figure 3a-c is a graphical representation of immunofluorescence intensities illustrating the effects of LPV, RTV and LPV/r on expression of BAX and BCL-2 in MCF-7 and MCF-10A cell cultures after 96 h of exposure. In the untreated group of MCF-7 cells (micrographs not shown), BAX and BCL-2 proteins were expressed and co-localised in the nuclei and cytoplasm. Mean fluorescence intensities of BAX and BCL-2 in the nuclei and cytoplasm were statistically analysed (ANOVA) between the untreated/vehicle control and the treated groups (graphical representation in Figure 3a-c).There are no differences in protein staining, localisation and intensity of vehicle (0.01% methanol) treated group when compared to growth medium alone. BAX and BCL-2, localisations remain unaltered across treatment groups. The untreated group of MCF-10A cells showed the expression and localisation of BAX and BCL-2 (micrographs not shown). Mean fluorescence intensities of BAX and BCL-2 in the nuclei and cytoplasm were statistically analysed (ANOVA) between the untreated (vehicle) control and the treated groups. (graphical representation in Figure 3a-c). There were no differences in protein staining, localisation and intensity of vehicle (0.01% methanol) treated group as compared to growth medium alone. BAX and BCL-2 localisations remain unchanged across treatment groups.
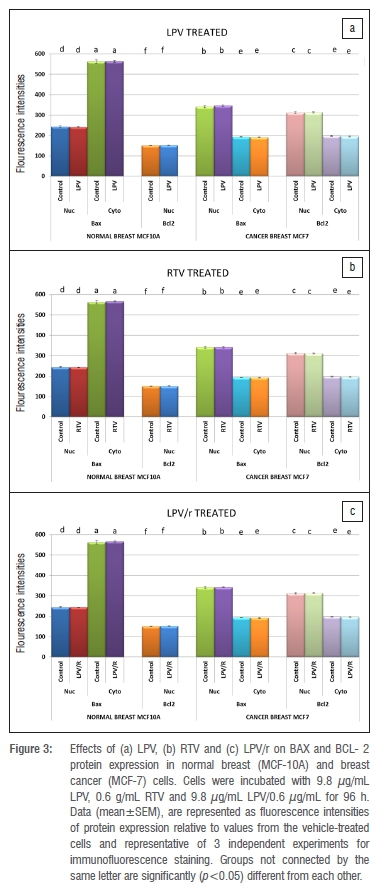
Protease inhibitors and apoptosis inhibition
Numerous studies19,20,32,39 have shown that protease inhibitors have potent anti-apoptotic effects in a variety of cell systems, the majority of which are immune cells. We assessed the anti-apoptotic effects of LPV/r to test whether these generalised effects are seen in the currently investigated human breast cell lines and to test whether they alter apoptosis via a pathway other than the BAX/BCL-2 pathway, since they had no significant effects on BAX and BCL-2 mRNA expression (Figure 2). From an evaluation of the many methods currently used to analyse apoptosis, chromatin condensation and nuclear fragmentation remain the hallmarks of apoptotic cells.30 Because it had been suggested that as a rule, classification of cell death in a given model should always include morphological examination coupled with at least one other assay,40 we chose the acridine orange staining for morphological evaluation and an ELISA based method for the quantitative analysis of DNA fragmentation. From previous optimisation experiments, it was determined that 1 μM and 30 μM CPT significantly induced apoptosis in the MCF-7 and MCF10-A cells respectively for 6 h. Figures 4 and 5 represent data from AO staining showing that a 96 h exposure to LPV/r did not significantly (p<0.05) inhibit apoptosis in both breast cell lines. Figure 6 represents data quantifying DNA fragmentation between groups. These ELISA results also confirm that LPV/r did not significantly inhibit drug-induced apoptosis in the breast MCF-7 and MCF-10A cells.
Discussion and conclusion
This study investigated the possibility that the use of protease inhibitors could be a risk factor for the initiation or development of breast cancer in patients receiving HAART. An analysis of the incidence of non-AIDS defining cancers in HIV-infected patients suggests that the incidence of NADM in these patients has significantly increased since the introduction and implementation of the combined antiretroviral therapy,12 creating the need to effectively establish or rule out the possibility that candidate antiretroviral drugs may promote breast cancer. The breast was chosen as a model of non-AIDS-defining malignancies.
The findings reported here show that the antiretroviral drugs: lopinavir, ritonavir and their combination (all at clinically relevant concentrations, which reflect their steady-state peak plasma concentrations in patients receiving these drugs)41,42 demonstrated some form of cytotoxicity in the breast cells, which may alter the activity of the nuclear and mitochondrial genome of cells and may cause genotoxic effects largely related to the tumour initiation processes.43-45 LPV showed an initial cytotoxic effect on the MCF-10A cells after 24 h of exposure. Even though this cytotoxic effect disappeared after 48 h, 72 h and 96 h of exposure, this initial cytotoxicity can produce genomic rearrangements that may act as the primary step toward carcinogenesis. RTV was significantly cytotoxic to the normal breast MCF-10A cells at 24 h, when compared with the control group and the breast cancer MCF-7 cells. The cytotoxic effects of RTV on the normal breast disappeared with continuous administration but could lead to DNA damage and may trigger the processes of cancer initiation in normal breast cells. RTV demonstrated opposing effects on normal breast and breast cancer cells at 24 h. It generally appeared to promote survival in normal breast after an initial toxic effect. Interestingly, the combination of the two protease inhibitors (lopinavir and ritonavir), as combined in Kaletra®, did not significantly alter the percentage viability of both breast cell lines.
To promote cancer, the antiretroviral drugs, either individually or in combination at the clinically relevant concentrations tested were expected to up-regulate the anti-apoptotic BCL-2 mRNA and or protein and down- regulate the pro-apoptotic BAX mRNA and or protein. Testing the effects of protease inhibitors (individually and in combination), our findings show that the antiretroviral drugs; lopinavir, ritonavir and Kaletra®, all at clinically relevant concentrations (which reflect their steady-state peak plasma concentrations), do not alter the mRNA expression of apoptosis related BAX and BCL-2. Neither do they alter the localisation of their proteins in the human breast cancer cell line MCF-7 and non-tumorigenic immortalised breast cell line MCF-10A. These results are consistent with the findings of other researchers. Reports by Gomez-Sucerquia et al.46 showed that efavirenz at similar concentrations did not alter the mRNA expression of BAX and BCL-2 like 1 genes following a 24 h treatment period in human Hep3B cells. Phenix et al.32, Badley19 and Rizza and Badley20 reported that protease inhibitors, including lopinavir, did not alter the mRNA expression and protein synthesis of BAX, BCL-2 and some other key apoptotic genes following a 3-day exposure in immune cells. Some other proteins and/or genes of this or another pathway, however, might be involved.
It has been shown previously that protease inhibitors have potent anti-apoptotic effects in different cellular systems, the majority of which are immune cells.19,20,32,39 These studies reported that the protease inhibitors investigated inhibited drug induced apoptosis. This anti-apoptotic property, (exhibited by protease inhibitors at concentrations similar to those levels achieved in people receiving the drugs) is an important phase in cancer development. The anti-apoptotic effect of LPV/r was therefore assessed to test whether these generalised effects are seen in the currently investigated cell lines and to test whether they alter apoptosis via a pathway other than the BAX/BCL-2 pathway, because they had no significant effects on BAX and BCL-2 mRNA expression. At a dose of 1 μM CPT significantly increased the percentage of apoptotic MCF-7 cells from 3-4% (spontaneous apoptosis) to 15-18% following a 6-h exposure, while the percentage of apoptotic MCF-10A cells increased from 1-2% to 8-9% following a 6 h exposure to 30 μM CPT. Pre-incubation with LPV/r did not significantly (p>0.05) inhibit CPT-induced apoptosis in both breast cell lines. The cell death ELISA assessment of DNA fragmentation showed similar results. These findings conflict with the reports of Phenix et al.32 which demonstrated that the HIV protease inhibitor nelfinavir inhibited Jurkat T-cell apoptosis induced by a variety of different stimuli, including CPT and with our previous report that pre-incubation with LPV/r for 96 h significantly inhibited the development of apoptosis in the cervical cancer HCS-2 cell line.47 These disparities might be because different cell lines react differently to the same target molecules. The variety of responses depends on the set of receptor proteins the cell possesses, which determines the specific subset of signals it can respond to, and also depends on the intracellular machinery by which the cell integrates and interprets the signals it receives.48 However, Phenix et al.32 and Adefolaju et al.47 also reported that inhibition of apoptotic death was not associated with alterations in mRNA expression of a variety of pro- and anti-apoptotic factors, and was not dependent on protein synthesis.
In the HAART era, it is a paradox that people living with HIV and AIDS are now at an increased risk for developing several specific non-AIDS-defining malignancies4,49-53 including breast cancer.12,52 Such that, despite the immune reconstitution induced by HAART, increased NADMs deaths have been reported amongst HIV patients.54 It has also been reported that patients with NADMs often have more aggressive cancers and display more advanced stages of the disease55,56, with breast cancer metastasising early and usually more poorly differentiated.57 The effects of HAART on the risk for NADM have not been clearly established. Research evidence has been conflicting as to whether or which antiretroviral drugs or classes decrease, increase, or have no effect on the risk of developing NADM,56,58 with no definite pattern emerging from such studies.
The speculation that some antiretroviral drugs or combinations may have an adverse effect on the risk of carcinogenesis among HIV patients under HAART led to this study. Our results show that these protease inhibitors do not alter the mRNA expression of apoptosis related BAX and BCL-2, neither do they alter the localisation of their proteins in the human breast cancer cell line MCF-7 and non-tumorigenic immortalised breast cell line MCF- 10A. The findings reported here also show that HIV protease inhibitors - LPV/r did not exhibit anti-apoptotic properties in MCF-7 and MCF-10A cells. If the ARVs initiate or promote cancer, these pathways are not likely to be involved, so the effects of these agents need to be investigated on other oncogenic pathways. Also, considering that this is an in-vitro study, it should be noted that the possibility of obtaining different results cannot be excluded when studied in an in vivo model, in which responses to pharmacological agents are much more complicated.
Acknowledgements
We thank Dr Clem Penny, Department of Internal Medicine, University of the Witwatersrand, Johannesburg, South Africa for providing the MCF- 7 and MCF-10A cells. This work was supported by the South African Medical Research Council.
Authors' contributions
G.A.A., K.E.T. and M.J.H. were involved with the conceptualisation of study and experimental design, standardisation and optimisation of all protocols, data acquisition, analysis and interpretation and manuscript write-up and final approval of the revised version. M.J.H was also involved with supervision of the project.
References
1. Joint United Nations Programme on HIV/AIDS (UNAIDS). Joint United Nations Programme on HIV/AIDS update on the global AIDS epidemic 2013. Geneva: UNAIDS; 2013. Available from: http://www.unaids.org/sites/default/files/en/media/unaids/contentassets/documents/ epidemiology/2013/ gr2013/UNAIDS_Global_Report_2013_en.pdf [ Links ]
2. Department of Health. The South African antiretroviral treatment guidelines 2010. Republic of South Africa. Pretoria: Department of Health; 2010. Available from: http://www.uj.ac.za/EN/CorporateServices/ioha/Documentation/Documents/ART%20Guideline.pdf [ Links ]
3. Nachega JB, Stein DM, Lehman DA, Hlatshwayo D, Mothopeng R, Chaisson RE, et al. Adherence to antiretroviral therapy in HIV-infected adults in Soweto, South Africa. AIDS Res Hum Retroviruses. 2004;20(10):1053-1056. [ Links ]
4. Sikora MJ, Rae JM, Johnson MD, Desta Z. Efavirenz directly modulates the oestrogen receptor and induces breast cancer cell growth. HIV Med. 2010;11(9):603-607. [ Links ]
5. Maqutu D, Zewotir T, North D, Naidoo K, Grobler A. Determinants of optimal adherence over time to antiretroviral therapy amongst HIV positive adults in South Africa: A longitudinal study. AIDS Behav. 2011;15(7):1465-1474. [ Links ]
6. Blas-Garcia A, Apostolova N, Ballesteros D, Monleon D, Morales JM, Rocha M, et al. Inhibition of mitochondrial function by efavirenz increases lipid content in hepatic cells. Hepatology. 2010;52(1):115-125. [ Links ]
7. Powles T, Robinson D, Stebbing J, Shamash J, Nelson M, Gazzard B, et al. Highly active antiretroviral therapy and the incidence of non-AIDS-defining cancers in people with HIV infection. J Clin Oncol. 2009;27(6):884-890. [ Links ]
8. Torres SM, Walker DM, Carter MM, Cook DL Jr, McCash CL, Cordova EM, et al. Mutagenicity of zidovudine, lamivudine, and abacavir following in vitro exposure of human lymphoblastoid cells or in utero exposure of CD-1 mice to single agents or drug combinations. Environ Mol Mutagen. 2007;48(3/4):224-238. [ Links ]
9. Oluwole SF, Ali AO, Shafaee Z, Depaz HA. Breast cancer in women with HIV/AIDS: Report of five cases with a review of the literature. J Surg Oncol. 2005;89(1):23-27. [ Links ]
10. Clay PG, Taylor TA, Glaros AG, McRae M, Williams C, McCandless D, et al. "One pill, once daily": What clinicians need to know about Atriplatrade mark. Ther Clin Risk Manag. 2008;4(2):291-302. [ Links ]
11. Phillips AA, Justman JE. Screening HIV-infected patients for non-AIDS-defining malignancies. Curr HIV/AIDS Rep. 2009;6(2):83-92. [ Links ]
12. Shiels MS, Cole SR, Kirk GD, Poole C. A meta-analysis of the incidence of non-AIDS cancers in HIV-infected individuals. J Acquir Immune Defic Syndr. 2009;52(5):611-622. [ Links ]
13. Clifford GM, Polesel J, Rickenbach M, Dal Maso L, Keiser O, Kofler A, et al. Cancer risk in the Swiss HIV Cohort Study: Associations with immunodeficiency, smoking, and highly active antiretroviral therapy. J Natl Cancer Inst. 2005;97(6):425-432. [ Links ]
14. Silverberg MJ, Abrams DI. AIDS-defining and non-AIDS-defining malignancies: Cancer occurrence in the antiretroviral therapy era. Curr Opin Oncol. 2007;19(5):446-451. [ Links ]
15. Engels EA, Biggar RJ, Hall HI, Cross H, Crutchfield A, Finch JL, et al. Cancer risk in people infected with human immunodeficiency virus in the United States. Int J Cancer. 2008;123(1):187-94. [ Links ]
16. Eum KH, Lee M. Crosstalk between autophagy and apoptosis in the regulation of paclitaxel-induced cell death in v-Ha-ras-transformed fibroblasts. Mol Cell Biochem. 2011;348(1/2):61-68. [ Links ]
17. Llambi F, Green DR. Apoptosis and oncogenesis: Give and take in the BCL-2 family. Curr Opin Genet Dev. 2011;21(1):12-20. [ Links ]
18. Sjostrom J, Bergh J. How apoptosis is regulated, and what goes wrong in cancer. BMJ. 2001;322(7301):1538-1539. [ Links ]
19. Badley AD. In vitro and in vivo effects of HIV protease inhibitors on apoptosis. Cell Death Differ. 2005;12:924-931. [ Links ]
20. Rizza SA, Badley AD. HIV protease inhibitors impact on apoptosis. Med Chem. 2008;4(1):75-79. [ Links ]
21. Razandi M, Alton G, Pedram A, Ghonshani S, Webb P Levin ER. Identification of a structural determinant necessary for the localization and function of estrogen receptor alpha at the plasma membrane. Mol Cell Biol. 2003;23(5):1633-1646. [ Links ]
22. Pan YWang L, Dai JL. Suppression of breast cancer cell growth by Na+/H+ exchanger regulatory factor 1 (NHERF1). Breast Cancer Res. 2006;8(6):R63. [ Links ]
23. Noor MA, Flint OP, Maa J-F, Parker RA. Effects of atazanavir/ritonavir and lopinavir/ritonavir on glucose uptake and insulin sensitivity: Demonstrable differences in vitro and clinically. AIDS. 2006;20(14):1813-1821. [ Links ]
24. Tong L, Phan TK, Robinson KL, Babusis D, Strab R, Bhoopathy S, et al. Effects of human immunodeficiency virus protease inhibitors on the intestinal absorption of tenofovir disoproxil fumarate in vitro. Antimicrob Agents Chemother. 2007;51(10):3498-3504. [ Links ]
25. Abbott Laboratories. Drug information: Norvir® [document on the Internet]. c2013 [cited 2015 Jan 15]. Available from: http://www.accessdata.fda.gov/drugsatfda_docs/label/2010/020945s028lbl.pdf [ Links ]
26. Abbott Laboratories. Drug information: Kaletra® [document on the Internet]. c2010 [cited 2015 Jan 15]. Available from: http://www.accessdata.fda.gov/drugsatfda_docs/label/2010/021226s030lbl.pdf [ Links ]
27. Borenfreund E, Puerner JA. Toxicity determined in vitro by morphological alterations and neutral red absorption. Toxicol Lett. 1985;24(2/3):119-124. [ Links ]
28. Hellemans J, Mortier G, De Paepe A, Speleman F, Vandesompele J. qBase relative quantification framework and software for management and automated analysis of real-time quantitative PCR data. Genome Biol. 2007;8(2):R19. [ Links ]
29. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25(4):402-408. [ Links ]
30. Ribble D, Goldstein NB, Norris DA, Shellman YG. A simple technique for quantifying apoptosis in 96-well plates. BMC Biotechnol. 2005;5:12. [ Links ]
31. Yang S, Liu M, Liang N, Zhao Q, Zhang YXue W, et al. Discovery and antitumor activities of constituents from Cyrtomium fortumei (J.) Smith rhizomes. Chem Cent J. 2013;7:24. [ Links ]
32. Phenix BN, Lum JJ, Nie Z, Sanchez-Dardon J, Badley AD. Antiapoptotic mechanism of HIV protease inhibitors: Preventing mitochondrial transmembrane potential loss. Blood. 2001;98(4):1078-1085. [ Links ]
33. Mironova EV, Evstratova AA, Antonov SM. A fluorescence vital assay for the recognition and quantification of excitotoxic cell death by necrosis and apoptosis using confocal microscopy on neurons in culture. J Neurosci Meth. 2007;163(1):1-8. [ Links ]
34. Canete M, Juarranz A, Lopez-Nieva P Alonso-Torcal C, Villanueva A, Stockert JC. Fixation and permanent mounting of fluorescent probes after vital labelling of cultured cells. Acta Histochem. 2001;103(2):117-126. [ Links ]
35. Liu CY, Takemasa A, Liles WC, Goodman RB, Jonas M, Rosen H, et al. Broad-spectrum caspase inhibition paradoxically augments cell death in TNF-alpha-stimulated neutrophils. Blood. 2003;101(1):295-304. [ Links ]
36. Tu HP Chen YT, Chiu HC, Chin YT, Huang SM, Cheng LC, et al. Cyclosporine A enhances apoptosis in gingival keratinocytes of rats and in OECM1 cells via the mitochondrial pathway. J Periodontal Res. 2009;44(6):767-775. [ Links ]
37. Nieves-Neira W, Pommier Y Apoptotic response to Camptothecin and 7-hydroxystaurosporine (UCN-01) in the 8 human breast cancer cell lines of the NCI anticancer drug screen: Multifactorial relationships with topoisomerase I, protein kinase C, Bcl-2, p53, MDM-2 and caspase pathways. Int J Cancer. 1999;82(3):396-404. [ Links ]
38. Rastogi S, Joshi B, Fusaro G, Chellappan S. Camptothecin induces nuclear export of prohibitin preferentially in transformed cells through a CRM-1-dependent mechanism. J Biol Chem. 2006;281(5):2951-2959. [ Links ]
39. Vlahakis SR, Bennett SA, Whitehead SN, Badley AD. HIV protease inhibitors modulate apoptosis signaling in vitro and in vivo. Apoptosis. 2007;12(5):969- 977. [ Links ]
40. Renvoize C, Biola A, Pallardy M, Breard J. Apoptosis: Identification of dying cells. Cell Biol Toxicol. 1998;14(2):111-120. [ Links ]
41. Apostolova N, Gomez-Sucerquia LJ, Moran A, Alvarez A, Blas-Garcia A, Esplugues JV. Enhanced oxidative stress and increased mitochondrial mass during efavirenz-induced apoptosis in human hepatic cells. Br J Pharmacol. 2010;160(8):2069-2084. [ Links ]
42. Bumpus NN. Efavirenz and 8-hydroxyefavirenz induce cell death via a JNK-and BimEL-dependent mechanism in primary human hepatocytes. Toxicol Appl Pharmacol. 2011;257(2):227-234. [ Links ]
43. Bishop AJ, Schiestl RH. Homologous recombination as a mechanism of carcinogenesis. Biochim Biophys Acta. 2001;1471(3):M109-121. [ Links ]
44. Olivero OA. Mechanisms of genotoxicity of nucleoside reverse transcriptase inhibitors. Environ Mol Mutagen. 2007;48(3/4):215-223. [ Links ]
45. Wu KM, Powley MW, Ghantous H. Timing of carcinogenicity studies and predictability of genotoxicity for tumorigenicity in anti-HIV drug development. Int J Toxicol. 2012;31(3):211-221. [ Links ]
46. Gomez-Sucerquia LJ, Blas-Garcia A, Marti-Cabrera M, Esplugues JV, Apostolova N. Profile of stress and toxicity gene expression in human hepatic cells treated with efavirenz. Antiviral Res. 2012;94(3):232-241. [ Links ]
47. Adefolaju GA, Theron KE, Hosie MJ. Effects of HIV protease, nucleoside/non-nucleoside reverse transcriptase inhibitors on Bax, Bcl-2 and apoptosis in two cervical cell lines. Biomed Pharmacother. 2014;68(2):241-251. [ Links ]
48. Alberts B, Johnson A, Lewis J, Raff, M, Roberts K, Walter P. Molecular biology of the cell. 4th ed. New York: Garland Science; 2002. [ Links ]
49. Silverberg MJ, Chao C, Leyden WA, Xu L, Tang B, Horberg MA, et al. HIV infection and the risk of cancers with and without a known infectious cause. AIDS. 2009;23(17):2337-2345. [ Links ]
50. Spano JP, Lanoy E, Mounier N, Katlama C, Costagliola D, Heard I. Breast cancer among HIV infected individuals from the ONCOVIH study, in France: Therapeutic implications. Eur J Cancer. 2012;48(18):3335-3341. [ Links ]
51. Calabresi A, Ferraresi A, Festa A, Scarcella C, Donato F, Vassallo F, et al. Incidence of AIDS-defining cancers and virus-related and non-virus-related non-AIDS-defining cancers among HIV-infected patients compared with the general population in a large health district of Northern Italy, 1999-2009. HIV Med. 2013;14(8):481-490. [ Links ]
52. Franzetti M, Adorni F, Parravicini C, Vergani B, Antinori S, Milazzo L, et al. Trends and predictors of non-AIDS-defining cancers in men and women with HIV infection: A single-institution retrospective study before and after the introduction of HAART. J Acquir Immune Defic Syndr. 2013;62(4):414-420. [ Links ]
53. Cutrell J, Bedimo R. Non-AIDS-defining cancers among HIV-infected patients. Curr HIV/AIDS Rep. 2013;10(3):207-216. [ Links ]
54. Zucchetto A, Suligoi B, De Paoli A, Pennazza S, Polesel J, Bruzzone S, et al. Excess mortality for non-AIDS-defining cancers among people with AIDS. Clin Infect Dis. 2010;51(9):1099-1101. [ Links ]
55. Grulich AE, Van Leeuwen MT, Falster MO, Vajdic CM. Incidence of cancers in people with HIV/AIDS compared with immunosuppressed transplant recipients: A meta-analysis. Lancet. 2007;370(9581):59-67. [ Links ]
56. Deeken JF, Tjen-A-Looi A, Rudek MA, Okuliar C, Young M, Little RF, et al. The rising challenge of non-AIDS-defining cancers in HIV-infected patients. Clin Infect Dis. 2012;55(9):1228-1235. [ Links ]
57. Gewurz BE, Dezube BJ, Pantanowitz L. HIV and the breast. AIDS Read. 2005;15(8):392-396, 399-102. [ Links ]
58. Burgi A, Brodine S, Wegner S, Milazzo M, Wallace MR, Spooner K, et al. Incidence and risk factors for the occurrence of non-AIDS-defining cancers among human immunodeficiency virus-infected individuals. Cancer. 2005;104(7):1505-1511. [ Links ]
 Correspondence:
Correspondence:
Gbenga Adefolaju
School of Anatomical Sciences, University of the Witwatersrand Medical School
7 York Road, Parktown 2193
South Africa
Email: Gbenga.Adefolaju@ul.ac.za
Received: 26 Nov. 2014
Revised: 19 Jan. 2015
Accepted: 09 Feb. 2015

