










Servicios Personalizados
Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkSouth African Journal of Science
versión On-line ISSN 1996-7489
versión impresa ISSN 0038-2353
S. Afr. j. sci. vol.110 no.1-2 Pretoria ene. 2014
REVIEW ARTICLE
Inflammation and cancer: The role of the human neutrophil
Ronald AndersonI; Gregory R. TintingerII; Charles FeldmanIII
IMedical Research Council Unit for Inflammation and Immunity, Department of Immunology, Faculty of Health Sciences, University of Pretoria, Pretoria, South Africa
IIDepartment of Internal Medicine, Steve Biko Academic Hospital, Faculty of Health Sciences, University of Pretoria, Pretoria, South Africa
IIIDivision of Pulmonology, Department of Internal Medicine, Charlotte Maxeke Johannesburg Academic Hospital, Faculty of Health Sciences, University of the Witwatersrand, Johannesburg, South Africa
ABSTRACT
Chronic inflammation of both infective and non-infective origin has been implicated in the aetiology of approximately 30% of all human epithelial malignancies. The primary carcinogens are reactive oxygen species (ROS) derived from activated, infiltrating cells of the innate immune system, especially neutrophils, which inflict oxidative damage on the DNA of bystander epithelial cells. The consequence is gene modifications which initiate cellular transformation. The process of tumourigenesis is exacerbated by the sustained generation of pro-proliferative ROS, as well as by the release of neutrophil-derived cytokines and proteases, all of which contribute to tumour promotion and progression. It is now well recognised that, in addition to inflammation causing cancer, many cancers per se induce an inflammatory response, with a high magnitude of neutrophil influx being indicative of a poor prognosis. In this setting, CXC chemokines produced by tumours not only promote neutrophil influx and hyperreactivity, but also cause autocrine activation of the proliferation of the chemokine-producing tumour cells. These various mechanisms of inflammation-mediated tumourigenesis are the primary focus of this review, together with a consideration of neutrophil-targeted anti-inflammatory strategies with potential as adjunctive cancer therapy.
Keywords: angiogenesis; chemokines; cytokines; hydrogen peroxide; proteinases; redox signalling; reactive oxygen species
Introduction
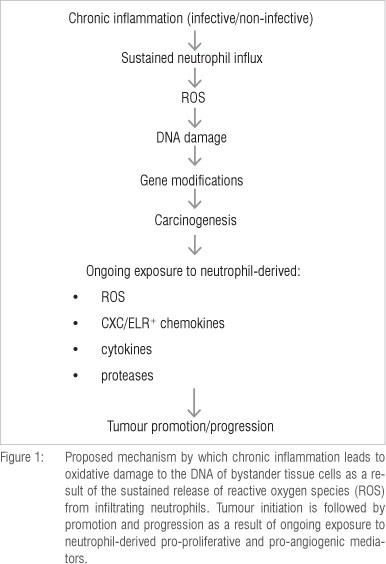
The link between cancer and inflammation was described 150 years ago by the distinguished German pathologist Rudolph Virchow.1,2 Based on epidemiological studies, it is now recognised that up to 30% of all cancers may have underlying inflammation-associated aetiology, triggered by chronic infection or other types of non-infective unresolved inflammation.3-6 In this setting, reactive oxygen and nitrogen species (ROS/RNS) released by activated phagocytes at sites of inflammation inflict oxidative damage on neighbouring bystander cells, especially epithelial cells, initiating tumourigenesis.7,8 Subsequent promotion/progression and metastasis involve not only ongoing oxidative stress, but also the release of pro-proliferative and pro-angiogenic/pro-metastatic, phagocyte-derived cytokines/chemokines and proteases.1-17
Although inflammation is a major and primary cause of cancer, many cancers per se also activate an inflammatory response, resulting in infiltration by various types of myeloid cells of the innate immune system. These cells include neutrophils, monocytes/macrophages, dendritic cells, and so-called myeloid-derived suppressor cells of both granulocytic and monocytic origin.18-23 Although this tumour-associated inflammatory response is potentially protective, at least in the case of neutrophils, monocytes/macrophages and dendritic cells,19,20 it may also contribute to tumour progression and metastasis through the mechanisms alluded to above.9
In this review, we focus on the pro-tumourigenic potential of the neutrophil, which, amongst other types of immune and inflammatory cells, is abundant in tumours, appearing to be an important, independent predictor of poor outcome in many,3,21 but not all, types24 of malignancy. The major themes addressed here are the roles of neutrophil-derived/-associated ROS, chemokines/cytokines, proteinases and adhesion molecules in tumour initiation, promotion, progression and metastasis, as well as the potential role of anti-inflammatory strategies in cancer prevention and therapy.
Neutrophils and carcinogenesis
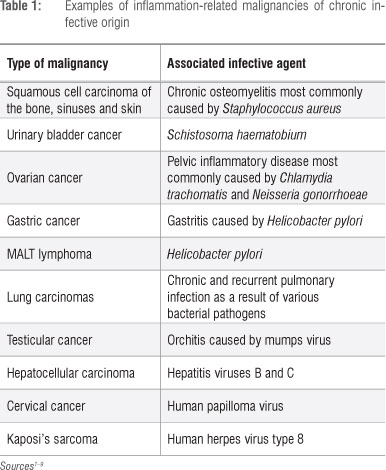
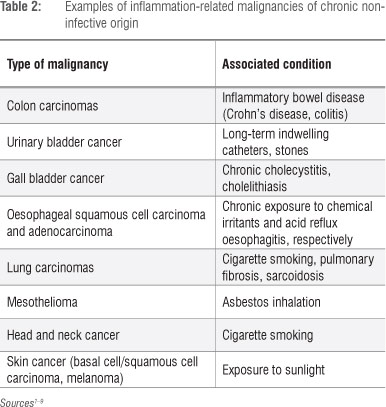
As described by Weitzman and Gordon in their seminal review7 and, more recently, by Knaapen et al.10, the propensity for cancers to develop at sites of inflammation is well recognised, the association being supported by compelling epidemiological and experimental evidence. Examples of inflammation-associated cancers, primarily epithelial, of both infective and non-infective origin are shown in Tables 1 and 2, respectively. In the setting of inflammation-associated cancer, phagocyte-derived ROS - produced and released extracellularly by infiltrating neutrophils - have been identified as the primary offenders.7,10 These indiscriminate, toxic agents are potent carcinogens, posing the potential hazard of oxidative damage to the DNA of bystander, host structural cells at sites of inflammation and resulting in the gene modifications which precede cellular transformation.
Convincing evidence demonstrating the carcinogenic potential of ROS was derived from experiments in which eukaryotic structural cells and lymphocytes were exposed to activated neutrophils, to cell-free enzymatic ROS-generating systems, or to the relatively stable, cell-permeant ROS hydrogen peroxide (H2O2) in vitro, which resulted in severe oxidative stress and damage to the genetic material of these cells.7,10 In all of these systems, direct oxidative damage to DNA appears to involve intracellular conversion of H2O2 to a highly potent and reactive ROS - hydroxyl radical - probably by Fenton-type mechanisms involving electron donation by heavy metals. The types of ROS-mediated damage include: (1) gross chromosomal damage (sister chromatid exchanges), (2) single- and double-DNA strand breaks and (3) oxidative damage to the bases in DNA.7,10 In the case of the latter, the signature of oxidative damage is conversion of guanosine to 8-hydroxydeoxyguanosine, although the other DNA bases are also vulnerable to oxidative damage.7 RNS produced predominantly by macrophages result in the formation of reactive aldehydes and malondialdehydes which also induce point mutations.8
In addition to the direct, DNA-damaging activities of phagocyte-derived ROS, these oxidants also inhibit the activities of several DNA repair enzymes, thereby exacerbating oxidative damage to genetic material.10 In this context, it is noteworthy that hypochlorous acid generated via the H2O2-dependent oxidation of chloride ions by myeloperoxidase (MPO), the neutrophil/monocyte primary granule enzyme, has been reported to interfere with the base excision repair enzyme poly (ADP-ribose) polymerase.25 Other DNA repair enzymes which are susceptible to oxidative inactivation include the glycolase Ogg1 and topoisomerase II, which are inactivated by nitric oxide and H2O2, respectively, compromising repair of 8-hydroxydeoxyguanosine moieties and strand scission/ligation.10,26,27
Clearly, ROS-mediated direct damage to DNA, together with oxidative dysfunction of DNA repair enzymes, predisposes to gene modifications,- which, particularly in the case of mutations to tumour suppressor and promoter genes, may lead to cellular transformation. However, these mechanisms are not the only ones by which phagocyte-derived ROS contribute to carcinogenesis. Other mechanisms include: (1) oxidative conversion of pre-carcinogenic chemicals/xenobiotics to complete carcinogens10,28, (2) redox activation of intracellular signalling mechanisms, which not only promote aberrant cellular proliferation, but also intensify inflammation-related oxidative stress29-34 and (3) oxidative suppression of the proliferative activity of anti-tumour T lymphocytes35,36. In addition to these mechanisms, several neutrophil-derived chemokines/ cytokines, proteinases and adhesion molecules also contribute to tumourigenesis via their pro-proliferative, pro-angiogenic and pro-metastatic activities.
Neutrophil-mediated oxidative activation of pre-carcinogens
Neutrophil-derived ROS, specifically those generated by the MPO/H2O2/halide system, have been implicated in the transformation to carcinogens of chemical pollutants generated by industrial, motor vehicle and household combustive processes, as well as those present in cigarette smoke.10 Examples of the former include aromatic and heterocyclic amines, especially polycyclic aromatic hydrocarbons, while benzo(a)pyrene in cigarette smoke undergoes oxidative conversion to BPDE (bay-region diol expoxides), which is mutagenic via formation of covalent adducts with guanine.
Redox activation of cellular proliferation
Unlike other ROS (such as superoxide anion, singlet molecular oxygen, hydroxyl radical and hypochlorous acid), H2O2 - because of its relative stability, cell permeability and ability to target proteins - can function efficiently as an intracellular signalling molecule.29-31 Indeed, it is well established that H2O2 can modulate cellular differentiation, proliferation, survival and synthesis of inflammatory mediators via the oxidative modification of key cysteine residues in various enzymes, including phosphatases and kinases, especially mitogen-activated protein kinases (MAPKs), as well as transcription factors.29-32 Under controlled conditions, H2O2 generated intracellularly in various types of cells by the ubiquitous, stringently regulated Nox (NADPH oxidase) family of enzymes, contributes positively to the maintenance of cellular homeostasis.29-32 However, when structural cells, especially epithelial cells, are subjected to intense oxidative stress, whether directly as a consequence of protracted activation of Nox enzymes or indirectly because of influx of extracellular H2O2 as a result of proximity to activated phagocytes, or both, then cell proliferation as a consequence of dysregulated intracellular signalling may ensue. Although disputed by those who believe that over-exposure to H2O2 is more likely to drive the cells into apoptosis,29-32 this scenario is countered to some extent by the following lines of evidence from experimental sources: (1) exposure of a Barrett's oesophagus adenocarcinoma cell line to low concentrations of H2O2 resulted in cell proliferation which was associated with sequential activation of extracellular regulated kinase 2, MAPK, the transcription factor, nuclear factor kappa B (NFkB) and Nox 5-S33; and (2) exposure of human oral cancer cells to the H2O2-producing microorganism Enterococcus faecalis resulted in catalase-inhibitable activation of the epidermal growth factor receptor and cell proliferation, underscoring the association between infection with this bacterial pathogen and oral carcinogenesis.34
ROS-mediated inactivation of tumour-targeted T cells
Although H2O2 at low concentrations can trigger the proliferation of epithelial cells via intracellular, redox signalling mechanisms, at higher concentrations this ROS can also promote the oxidative inactivation of the protective activities of T lymphocytes.35,36 In the setting of murine models of experimental tumourigenesis, infiltrating phagocytes, most notably a subset of activated neutrophils, have been found to inhibit the protective responses of tumour-targeted T-lymphocytes. The mechanism of immunosuppression involves intimate cell-cell contact mediated by the neutrophil p2-integrin Mac-1, exposing the T cells to high concentrations of neutrophil-derived H2O2.35,36
Neutrophil-derived cytokines in tumourigenesis
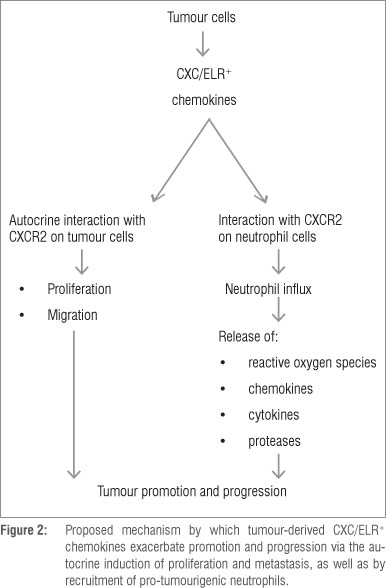
Although originally believed to have a very short lifespan and an extremely limited biosynthetic capacity, the survival time of neutrophils in the circulation of healthy humans has recently been reported to be 5.4 days.37 Following extravasation to sites of infection, tissue injury or cancer, this time may be considerably longer because of exposure to anti-apoptotic cytokines, especially granulocyte colony-stimulating factor (G-CSF) and granulocyte/macrophage colony-stimulating factor (GM-CSF).38 Extended survival of neutrophils is associated with acquisition of the capacity, albeit limited, to synthesise cytokines or chemokines,14,39 some of which are already stored in pre-synthesised, rapidly mobilisable form in cytoplasmic secondary and tertiary granules.40,41 These include: (1) the pro-angiogenic growth factor vascular endothelial growth factor (VEGF), (2) the chemokine interleukin (IL)-8 and the cytokines IL-1 p, IL-6 and TNF, all of which interact to promote neutrophil extravasation, accumulation and activation and (3) IL-12 which links innate and adaptive immunity, promoting cell-mediated immune responses involving T helper 1 lymphocytes.14,39-41 With respect to tumour promotion/progression, the most prominent of these are VEGF, which promotes tumour neovascularisation, and IL-8, which not only sustains neutrophil influx and activation, but also promotes tumour cell proliferation by the autocrine and paracrine mechanisms described under 'Chemokines and tumourigenesis'.
Although the evidence is somewhat less compelling than that for VEGF and IL-8, several other neutrophil-derived cytokines have been implicated in tumour promotion/progression and angiogenesis. Because these have been extensively reviewed recently,14 they are considered only briefly here.
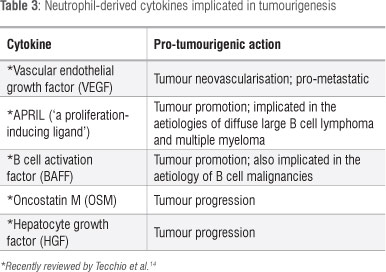
APRIL (also known as 'a proliferation-inducing ligand') and BAFF (B cell activation factor, BLyS), both of which belong to the TNF ligand family, interact with several receptors, especially BMCA (B cell maturation antigen), TAC1 and BAFF receptor, inducing B cell proliferation and survival. Both of these cytokines are produced by tumour-infiltrating neutrophils, and have been implicated in tumour promotion in malignancies such as diffuse large B cell lymphoma and multiple myeloma.14
Oncostatin M (OSM) and hepatocyte growth factor (HGF) are cytokines which are both stored and synthesised by tumour-infiltrating neutrophils. OSM appears to mediate tumour progression via induction of detachment of tumour cells and activation of synthesis of pro-angiogenic VEGF and fibroblast growth factor by endothelial cells, while HGF induces an invasive phenotype.14
These various cytokines and their reported roles in tumourigenesis are summarised in Table 3, with the exception of IL-8 which is discussed in detail below.
Chemokines and tumourigenesis
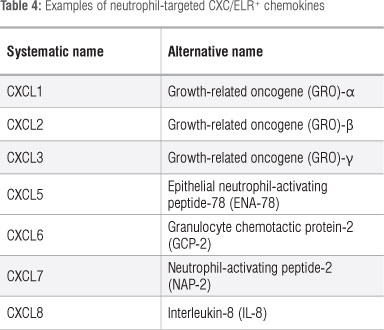
Chemokines are a group of low molecular weight chemotactic cytokines which promote the receptor-mediated migration of cells of the innate and adaptive immune systems. In the case of neutrophils, these cells are attracted to sites of tissue injury or infection by members of the sub-family of CXC/ELR-motif-positive chemokines (CXC denotes the presence of an intervening amino acid, X, between the first two conserved cysteine residues, while the ELR motif is a glu-leu-arg sequence preceding the first conserved cysteine residue). The various members of this chemokine sub-family are shown in Table 4, with the potent neutrophil chemoattractant IL-8 (CXCL8) predominating.
Notwithstanding production by cells of the innate and adaptive immune systems, CXC/ELR+ chemokines, especially IL-8, are also produced by various types of structural cells, including epithelial and endothelial cells, fibroblasts and smooth muscle cells. The major counter-receptor for these CXC/ELR+ chemokines is CXCR2 (IL-8 also interacts with CXCR1), which is expressed not only on neutrophils and mast cells, but also on epithelial and endothelial cells.12
Importantly, and aside from their primary role in neutrophil mobilisation, CXC/ELR+ chemokines and CXCR2 are also expressed by a diverse range of human cancers, including cancers of the breast, bladder, cervix, colon, liver, lymphatics, oesophagus, ovary, prostate and skin.12,13,42 In this setting, these chemokines drive tumour expansion via both autocrine and paracrine pro-proliferative interactions with CXCR2-expressing tumour cells.12,13,42,43 In the case of oesophageal squamous epithelial cells, and probably other tumour cell types, CXCR2-mediated proliferation results from activation of the transcription factor early growth response-1.11 In addition, tumour neovascularisation is mediated via the pro-angiogenic activities of these chemokines, especially IL-8,12 while the chronic influx of inflammatory cells exacerbates ROS-mediated oxidative damage to DNA and immunosuppression.
Neutrophil-derived proteases in tumour angiogenesis and metastasis
Neutrophil-derived proteases - specifically elastase and matrix metallo-proteinase-9 (MMP-9) stored in primary and secondary/tertiary granules, respectively - have also been implicated in inflammation-associated tumour neovascularisation and invasion. Elastase has been reported to degrade the intercellular adhesion molecule cadherin,44 while MMP-9 is a potent inducer of angiogenesis and tumour metastasis.16,17
Neutrophil adhesion molecules in tumour metastasis
Notwithstanding the expression of counter-receptors for endothelial adhesion molecules by some types of tumours,2 pro-adhesive interactions between circulating neutrophils, albeit in an animal model of experimental liver metastasis, have also been reported to mediate delivery of tumour cells to distant sites.45 In this setting, neutrophil/tumour cell adhesion is mediated via interactions of the [32-integrin Mac-1 on neutrophils, with its counter-receptor, intercellular adhesion molecule-1 (ICAM-1), on tumour cells.45
The aforementioned mechanisms of neutrophil/inflammation-mediated tumourigenesis are summarised in Figures 1 and 2.
Inflammation-targeted chemotherapy and immunotherapy in cancer
The chemopreventive potential of aspirin in particular, and possibly other non-steroidal anti-inflammatory drugs (NSAIDs), in reducing the incidence of colorectal cancer, and possibly other cancers, such as those of the liver, lung, oesophagus and stomach, is well recognised.2,19,46 - 48 Although the underlying mechanism is presumed to be anti-inflammatory in origin, other mechanisms, such as attenuation of prostaglandin E2-mediated inhibition of tumour-targeted T lymphocytes, have also been proposed.2 In the case of therapy, the potential of NSAIDs as adjuncts to conventional anti-cancer therapies remains largely unknown, a possible exception being the use of aspirin in the treatment of colorectal cancer associated with PIK3CA gene mutations.49,50
Other potential pharmacological strategies include the use of inhibitors of MMP-9, although these have proved disappointing in phase II/III clinical trials in various types of malignancy,51 and, perhaps the most promising strategy albeit unproven in the clinical setting, the use of pharmacological antagonists of CXCR243 and possibly dual antagonists of CXCR1/CXCR2. In addition to these, other categories of pharmacological agent which target the pro-inflammatory activities of neutrophils include 14/15-membered macrolide antibiotics and inhibitors of type 4 phosphodiesterase (PDE), the predominant PDE in human neutrophils. Unlike corticosteroids, which have limited efficacy in controlling neutrophilic inflammation, macrolides and PDE4 inhibitors possess a range of neutrophil-targeted anti-inflammatory activities- which have recently been described in detail elsewhere.52 Although untested with respect to their adjunctive potential as anti-inflammatory agents in cancer chemotherapy, it is noteworthy that novel macrolides and PDE4 inhibitors are currently under investigation for their direct anti-tumour activities.53,54
Monoclonal antibody-based anti-inflammatory therapies include those which target VEGF in various types of metastatic cancer, a strategy which has enjoyed variable success.55-57 Although monoclonal antibodies which target neutrophil-mobilising cytokines such as TNF, IL-8 and, more recently, IL-17A58,59 have been proposed as adjunctive anti-inflammatory strategies in the therapy of cancer, inhibitors of CXCR2 appear to be a superior option.
Conclusions
Although they are key players in innate host defence, human neutrophils are also inadvertent participants in the aetiology of inflammation-related cancers via the release of carcinogenic ROS and other mediators which contribute to tumour promotion and progression. Other types of cancer, which are not inflammatory in origin, also utilise inflammatory mechanisms to enhance their proliferative and invasive potential. The most significant of these mechanisms is the production of CXC/ELR+ chemokines. These chemokines not only recruit pro-tumourigenic neutrophils, but are also pro-proliferative and pro-metastatic via their autocrine interactions with CXCR2 expressed on tumour cells. These important insights into inflammation-associated mechanisms of tumourigenesis have enabled identification of potential anti-inflammatory adjunctive strategies to complement conventional anti-cancer therapies. However, given the range of neutrophil- and tumour-derived inflammatory mediators which contribute to tumourigenesis, selective targeting of a single mediator is unlikely to be successful. Although unproven in the clinical setting, selective antagonists of CXCR2, which target both neutrophils and tumour cells, represent a possible exception, as do NSAIDs, particularly aspirin. Preventive strategies include routine intake of low-dose NSAIDs, immunisation against cancer-causing viral pathogens, early aggressive antimicrobial chemotherapy to eradicate chronic inflammation caused by microbial pathogens, and avoidance of pro-inflammatory aspects of lifestyle such as cigarette smoking and excessive exposure to ultraviolet radiation.
Authors' contributions
All authors were involved in the overall conception, planning, design and writing of the manuscript.
References
1. Balkwill F, Mantovani A. Inflammation and cancer: Back to Virchow. Lan-cet.2001;357(9255):539-545. http://dx.doi.org/10.1016/S0140-6736(00) 04046-0 [ Links ]
2. Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420(6917):860-867. http://dx.doi.org/10.1038/nature01322 [ Links ]
3. Amulic B, Cazalet C, Hayes GL, Metzier KD, Zychlinsky A. Neutrophil function: From mechanisms to disease. Annu Rev Immunol. 2012;30:459-489. http://dx.doi.org/10.1146/annurev-immunol-020711-074942 [ Links ]
4. Chaturvedi AK, Gaydos CA, Agreda P Holden JP Chatterjee N, Goedert JJ, et al. Chlamydia pneumoniae infection and risk for lung cancer. Cancer Epidemiol Biomarkers Prev. 2010;19(6):1498-1505. http://dx.doi.org/10.1158/1055-9965.EPI-09-1261 [ Links ]
5. Shebl FM, Engels EA, Goedert JJ, Chaturvedi AK. Pulmonary infections and risk of lung cancer among persons with AIDS. J Acquir Immune Defic Syndr. 2010;55(3):375-379. http://dx.doi.org/10.1097/QAI.0b013e3181eef4f7 [ Links ]
6. Shiels MS, Albanes D, Virtamo J, Engels EA. Increased risk of lung cancer in men with tuberculosis in the alpha-tocopherol, beta-carotene cancer prevention study. Cancer Epidemiol Biomarkers Prev. 2011;20(4):672-678. http://dx.doi.org/10.1158/1055-9965.EPI-10-1166 [ Links ]
7. Weitzman SA, Gordon LI. Inflammation and cancer: Role of phagocyte-generated oxidants in carcinogenesis. Blood. 1990;76(4):655-663. [ Links ]
8. Perwez Hussain S, Harris CC. Inflammation and cancer: An ancient link with novel potentials. Int J Cancer. 2007;121(11):2373-2380. http://dx.doi.org/10.1002/ijc.23173 [ Links ]
9. Rakoff-Nahoum S. Why cancer and inflammation. Yale J Biol Med. 2006;79(3-4):123-130. [ Links ]
10. Knaapen AM, Güngõr N, Schins RPF, Borm PJA, Van Schooten FJ. Neutrophils and respiratory tract DNA damage and mutagenesis: A review. Mutagenesis. 2006;21(4):225-236. http://dx.doi.org/10.1093/mutage/gel032 [ Links ]
11. Wang B, Khachigian LM, Esau L, Birrer MJ, Zhao X, Parker MI, et al. A key role for early growth response-1 and nuclear factor-KB in mediating and maintaining GRO/CXCR2 proliferative signaling in esophageal cancer. Mol Cancer Res. 2009;7(5):755-764. http://dx.doi.org/10.1158/1541-7786.MCR-08-0472 [ Links ]
12. Verbeke H, Struyf S, Laureys G, Van Damme J. The expression and role of CXC chemokines in colorectal cancer. Cytokine Growth Factor Rev. 2011;22(5-6):345-358. http://dx.doi.org/10.1016/j.cytogfr.2011.09.002 [ Links ]
13. Verbeke H, Karel G, Van Damme J, Struyf S. The role of CXC chemokines in the transition of chronic inflammation to esophageal and gastric cancer. Biochim Biophys Acta. 2012;1825(1):117-129. [ Links ]
14. Tecchio C, Scapini P Pizzolo G, Cassatella MA. On the cytokines produced by human neutrophils in tumors. Sem Cancer Biol. 2013;23(3):159-170. http://dx.doi.org/10.1016/j.semcancer.2013.02.004 [ Links ]
15. Shimizu M, Tanaka T, Moriwaki H. Obesity and hepatocellular carcinoma: Targeting obesity-related inflammation for chemoprevention of liver carcinogen-esis. Semin Immunopathol. 2013;35(2):191-202. http://dx.doi.org/10.1007/s00281-012-0336-6 [ Links ]
16. Ardi VC, Kupriyanova TA, Deryugina EI, Quigley JP. Human neutrophils uniquely release TIMP-free MMP-9 to provide a potent catalytic stimulator of angiogenesis. Proc Natl Acad Sci USA. 2007;104(51):20262-20267. http://dx.doi.org/10.1073/pnas.0706438104 [ Links ]
17. Bekes EM, Schweighofer B, Kupriyanova TA, Zajac E, Ardi VC, Quigley JP et al. Tumor-recruited neutrophils and neutrophil TIMP-free MMP-9 regulate coordinately the levels of tumor angiogenesis and efficiency of malignant cell intravasation. Am J Pathol. 2011;179(3):1455-1470. http://dx.doi.org/10.1016/j.ajpath.2011.05.031 [ Links ]
18. Dumitru CA, Lang S, Brandau S. Modulation of neutrophil granulocytes in the tumor microenvironment: Mechanisms and consequences for tumor progression. Sem Cancer Biol. 2013;23(3):141-148. http://dx.doi.org/10.1016/j.semcancer.2013.02.005 [ Links ]
19. Gregory AD, Houghton AM. Tumor-associated neutrophils: New targets for cancer therapy. Cancer Res. 2011;71(7):2411-2416. http://dx.doi.org/10.1158/0008-5472.CAN-10-2583 [ Links ]
20. Brandau S, Dumitru CA, Lang S. Protumor and antitumor functions of neutrophil granulocytes. Semin Immunopathol. 2013;35(2):163-176. http://dx.doi.org/10.1007/s00281-012-0344-6 [ Links ]
21. Brandau S. The dichotomy of neutrophil granulocytes in cancer. Sem Cancer Biol. 2013;23(3):139-140. http://dx.doi.org/10.1016/j.semcan-cer.2013.02.008 [ Links ]
22. Toh B, Abastado J-P. Myeloid cells: Prime drivers of tumor progression. On-coImmunol. 2012;1(8):1360-1367. http://dx.doi.org/10.4161/onci.22196 [ Links ]
23. Evans A, Costello E. The role of inflammatory cells in fostering pancreatic cancer cell growth and invasion. Frontiers Physiol. 2012;3(270):1-7. [ Links ]
24. Carus A, Ladekari M, Hager H, Pilegaard H, Nielsen PS, Donskov F. Tumor-associated neutrophils and macrophages in non-small cell lung cancer: No immediate impact on patient outcome. Lung Cancer. 2013;81(1):130-137. http://dx.doi.org/10.1016/jJungcan.2013.03.003 [ Links ]
25. Van Rensburg CE, Van Staden AM, Anderson R. Inactivation of poly (ADP-ribose) polymerase by hypochlorous acid. Free Radic Biol Med. 1991;11(3):285-291. http://dx.doi.org/10.1016/0891-5849(91)90125-M [ Links ]
26. Jaiswal M, La Russo NF, Nishioka N, Nakabeppu Y Gores GJ. Human Ogg1, a protein involved in the repair of 8-oxoguanine, is inhibited by nitric oxide. Cancer Res. 2001;61(17):6388-6393. [ Links ]
27. Cai YJ, Lu JJ, Zhu H, Xie H, Huang M, Lin LP et al. Salvicine triggers DNA double-strand breaks and apoptosis by GSH-depletion-driven H2O2 generation and topoisomerase II inhibition. Free Radic Biol Med. 2008;45(5):627-635. http://dx.doi.org/10.1016/j.freeradbiomed.2008.05.017 [ Links ]
28. Vlasova II, Feng W-H, Goff JP Giorgianni A, Do D, Gollin SM, et al. Mye-loperoxidase-dependent oxidation of etoposide in human myeloid progenitor CD34+ cells. Mol Pharmacol. 2011;79(3):479-487. http://dx.doi.org/10.1124/mol.110.068718 [ Links ]
29. Forman HJ, Maiorino M, Ursini F. Signaling functions of reactive oxygen species. Biochemistry. 2010;49(5):835-842. http://dx.doi.org/10.1021/ bi9020378 [ Links ]
30. Gough DR, Cotter TG. Hydrogen peroxide: A Jekyll and Hyde signalling molecule. Cell Death Dis. 2011;2:e213. http://dx.doi.org/10.1038/cddis.2011.96 [ Links ]
31. Veal E, Day A. Hydrogen peroxide as a signaling molecule. Antioxid Redox Signal. 2011;15(1):147-151. http://dx.doi.org/10.1089/ars.2011.3968 [ Links ]
32. Runchel C, Matsuzawa A, Ichijo H. Mitogen-activated protein kinases in mammalian oxidative stress responses. Antioxid Redox Signal. 2011;15(1):205-218. http://dx.doi.org/10.1089/ars.2010.3733 [ Links ]
33. Zhou X, Li D, Resnick MB, Behar J, Wands J, Cao W. Signaling in H2O2-induced increase in cell proliferation in Barrett's esophageal adenocarcinoma cells. J Pharmacol Exp Ther. 2011;339(1):218-227. http://dx.doi.org/10.1124/jpet.111.182352 [ Links ]
34. Boonanantanasarn K, Gill AL, Yap Y Jayaprakash V Sullivan MA, Gill SR. Enterococcus faecalis enhances cell proliferation through hydrogen peroxide mediated epidermal growth factor receptor activation. Infect Immun. 2012;80(10):3545-3558. http://dx.doi.org/10.1128/IAI.00479-12 [ Links ]
35. Kusmartsev S, Nefedova Y, Yoder D, Gabrilovich DI. Antigen-specific inhibition of CD8+ T cell response by immature myeloid cells in cancer is mediated by reactive oxygen species. J Immunol. 2004;172(2):989-999. [ Links ]
36. Pillay J, Kamp VM, Van Hoffen E, Visser T, Tak T, Lammers JW, et al. A subset of neutrophils in human systemic inflammation inhibits T cell responses through Mac-1. J Clin Invest. 2012;122(1):327-336. http://dx.doi.org/10.1172/JCI57990 [ Links ]
37. Pillay J, Den Braber I,Vrisekoop N, Kwast LM, De Boer RJ, Borghans JA, et al. In vivo labeling with 2H2O reveals a human neutrophil lifespan of 5.4 days. Blood. 2010;116(4):625-627. http://dx.doi.org/10.1182/blood-2010-01-259028 [ Links ]
38. Milot E, Filep JG. Regulation of neutrophil survival/apoptosis by Mcl-1. Scientific World J. 2011;11:1948-1962. http://dx.doi.org/10.1100/2011/131539 [ Links ]
39. Cassatella MA. Neutrophil-derived proteins: Selling cytokines by the pound. Adv Immunol. 1999;73:369-509. http://dx.doi.org/10.1016/S0065-2776(08)60791-9 [ Links ]
40. Gaudry M, Brégerie O, Andrieu V Benna JE, Pocidalo M-A, Hakim J. Intracel-lular pool of vascular endothelial growth factor in human neutrophils. Blood. 1997;90(10):4153-4161. [ Links ]
41. Lacy P Stow JL. Cytokine release from innate immune cells: Association with diverse membrane trafficking pathways. Blood. 2011;118(1):9-18. http://dx.doi.org/10.1182/blood-2010-08-265892 [ Links ]
42. Lazennec G, Richmond A. Chemokines and chemokine receptors: New insights into cancer-related inflammation. Trends Mol Med. 2010;16(3):133-144. http://dx.doi.org/10.1016/j.molmed.2010.01.003 [ Links ]
43. Jamieson T, Clarke M, Steele CW, Samuel MS, Neumann J, Jung A, et al. Inhibition of CXCR2 profoundly suppresses inflammation-driven and spontaneous tumorigenesis. J Clin Invest. 2012;122(9):3127-3144. http://dx.doi.org/10.1172/JCI61067 [ Links ]
44. Grosse-Steffen T, Giese T, Giese N, Longerich T, Schirmacher P Hãnsch GM, et al. Epithelial-to-mesenchymal transition in pancreatic tumor cell lines: The role of neutrophils and neutrophil-derived elastase. Clin Dev Immunol. 2012;2012:720768. http://dx.doi.org/10.1155/2012/720768 [ Links ]
45. Spicer JD, McDonald B, Cools-Lartigue JJ, Chow SC, Giannias B, Kubes P et al. Neutrophils promote liver metastasis via Mac-1-mediated interactions with circulating tumor cells. Cancer Res. 2012;72(16):3919-3927. http://dx.doi. org/10.1158/0008-5472.CAN-11-2393 [ Links ]
46. Vandramini-Costa DB, Carvalho JE. Molecular link mechanisms between inflammation and cancer. Curr Pharm Des. 2012;18(26):3831-3852. http://dx.doi.org/10.2174/138161212802083707 [ Links ]
47. Levy IG, Pim CP. An aspirin a day: The allure (and distraction) of chemo-prevention. J Natl Cancer Inst. 2012;104(23):1782-1784. http://dx.doi.org/10.1093/jnci/djs462 [ Links ]
48. Sahasrabuddhe VV Gunja MZ, Graubard BI, Trabert B, Schwartz LM, Park Y et al. Non-steroidal anti-inflammatory drug use, chronic liver disease, and hepatocellular carcinoma. J Natl Cancer Inst. 2012;104(23):1808-1814. http://dx.doi.org/10.1093/jnci/djs452 [ Links ]
49. Liao X, Lochhead P, Nishihara R, Morikawa T, Kuchiba A, Yamauchi M, et al. Aspirin use, tumor PIK3CA mutation, and colorectal cancer survival. N Engl J Med. 2012;367(17):1596-1606. http://dx.doi.org/10.1056/NEJ-Moa1207756 [ Links ]
50. Pasche B. Aspirin - from prevention to targeted therapy. N Engl J Med. 2012;367(17):1650-1651. http://dx.doi.org/10.1056/NEJMe1210322 [ Links ]
51. Zuker S, Cao J. Selective matrix metalloproteinase (MMP) inhibitors in cancer therapy: Ready for prime time? Cancer Biol Ther. 2009;8(24):2371-2373. http://dx.doi.org/10.4161/cbt.8.24.10353 [ Links ]
52. Tintinger GR, Anderson R, Feldman C. Pharmacological approaches to regulate neutrophil activity. Semin Immunopathol. 2013;35(4):395-409. http://dx.doi.org/10.1007/s00281-013-0366-8 [ Links ]
53. Napolitano JG, Daranas AH, Norte M, Fernandez JJ. Marine macrolides, a promising source of antitumor compounds. Anti-Cancer Agents Med Chem. 2009;9(2):122-137. http://dx.doi.org/10.2174/187152009787313800 [ Links ]
54. Sengupta R, Sun R, Warrington NM, Rubin JB. Treating brain tumors with PDE4 inhibitors. Trends Pharmacol Sci. 2011;32(6):337-344. http://dx.doi.org/10.1016/j.tips.2011.02.015 [ Links ]
55. Wagner AD, Thomssen C, Haerting J, Unverzagt S. Vascular-endothelial-growth-factor (VEGF) targeting therapies for endocrine refractory or resistant metastatic breast cancer. Cochrane Database Syst Rev. 2012;7:CD008941. [ Links ]
56. Gianni L, Romieu GH, Lichinitser M, Serrano SV, Mansutti M, Pivot X, et al. AVEREL: A randomized phase III trial evaluating bevacizumab in combination with docetaxel and trastuzumab as first-line therapy for HER2-positive locally recurrent/metastatic breast cancer. J Clin Oncol. 2013;31(14):1719-1725. http://dx.doi.org/10.1200/JCO.2012.44.7912 [ Links ]
57. Makhoul I, Klimberg VS, Korourian S, Henry-Tillman RS, Siegel ER, West-brook KC, et al. Combined neoadjuvant chemotherapy with bevacizumab improves pathologic complete response in patients with hormone receptor negative operable or locally advanced breast cancer. Am J Clin Oncol. In press 2013. http://dx.doi.org/10.1097/COC.0b013e31828940c3 [ Links ]
58. Charles KA, Kulbe H, Soper R, Escorcio-Correia M, Lawrence T, Schultheis A, et al. The tumor-promoting actions of TNF-alpha involve TNFR1 and IL-17 in ovarian cancer in mice and humans. J Clin Invest. 2009;119(10):3011-3023. http://dx.doi.org/10.1172/JCI39065 [ Links ]
59. Oshiro K, Kohama H, Umemura M, Uyttenhove C, Inagaki-Ohara K, Arakawa T, et al. Interleukin-17A is involved in enhancement of tumor progression in murine intestine. Immunobiology. 2012;217(1):54-60. http://dx.doi.org/10.1016/j.imbio.2011.08.002 [ Links ]
 Correspondence:
Correspondence:
Ronald Anderson
Department of Immunology
University of Pretoria
PO Box 2034, Pretoria 0001, South Africa
Email: ronald.anderson@up.ac.za
Received: 02 July 2013
Revised: 12 Sep. 2013
Accepted: 12 Sep. 2013
© 2014. The Authors. Published under a Creative Commons Attribution Licence.

